Ce documentaire aborde en détail les différents mythes et procédures sur lesquels reposent la théorie scientifique des germes pathogènes, prétendument à l’origine de phénomènes de contagion, aussi appelée théorie des germes ou théorie microbienne.
L’histoire des épidémies telle que la poliomyélite, la variole et la grippe espagnole est retracée et le jargon des virologues et leurs techniques comme la PCR, le séquençage, l’effet cytopathique, le microscope électronique, les cultures cellulaires, l’isolement, la purification ou encore le concept d’anticorps sont analysés pour mieux comprendre sur quoi repose la virologie moderne et la théorie des germes dans son ensemble.
Le Dr Michael Yeadon, ancien directeur scientifique et vice-président de Pfizer, confirme sa conviction que les virus respiratoires n’existent pas.
J’ai réalisé, au fil du temps, que je ne pouvais plus maintenir ma compréhension des virus respiratoires telle que je pensais les connaître. Et puis j’ai appris de nouvelles informations récemment, et c’était juste, ça a effondré la possibilité que les virus respiratoires tels que décrits existent, ils n’existent pas.
Cette étude très influente, publiée pour la première fois en 2006, a contribué à orienter des milliards de dollars de recherche sur la maladie.
Selon un rapport publié dans Science, des éléments essentiels de l’un des travaux de recherche sur la maladie d’Alzheimer les plus cités au cours des deux dernières décennies pourraient avoir été délibérément manipulés.
La maladie d’Alzheimer est la cause la plus courante de démence dans le monde, selon l’Organisation mondiale de la santé. L’article très influent, publié dans Nature en 2006, a contribué à orienter des milliards de dollars de recherche du gouvernement américain sur la maladie d’Alzheimer, selon Science.
L’étude, qui portait sur le déclin cognitif chez les souris, proposait qu’une protéine amyloïde spécifique puisse être responsable du déclin cognitif. L’hypothèse a depuis lors dominé le débat, et les chercheurs s’efforcent depuis des années de comprendre le mécanisme par lequel ces protéines peuvent entraîner le déclin.
Mais un neuroscientifique du Tennessee, Matthew Schrag, professeur à l’université Vanderbilt, a déclaré dans un article de Science que lui et d’autres examinateurs ont identifié jusqu’à 10 articles sur la protéine qui méritent un examen plus approfondi. Le rapport cite également d’autres chercheurs éminents qui ont eu des difficultés à reproduire les résultats des études sur les protéines spécifiques.
“Je me concentre sur ce que nous pouvons voir dans les images publiées, et je les décris comme des signaux d’alarme, et non comme des conclusions finales”, a-t-il déclaré à Science, en révélant son rôle de lanceur d’alerte. “Les données devraient parler d’elles-mêmes”.
Le cœur du problème est de savoir si les images figurant dans plusieurs articles ont été manipulées pour mieux étayer une hypothèse, le travail du chercheur Sylvain Lesné faisant l’objet d’un examen particulier. Lesné, professeur associé à l’université du Minnesota, fait l’objet d’une enquête de la part de l’université.
Son coauteur sur plusieurs articles est le Dr Karen Ashe, également chercheur à l’université du Minnesota et l’un des plus éminents chercheurs sur la maladie d’Alzheimer du pays. Elle a décrit la manipulation potentielle des images comme étant “dévastatrice”, au Minneapolis Star Tribune, mais a critiqué l’idée selon laquelle ses recherches sur les protéines amyloïdes ont singulièrement orienté les dépenses du gouvernement et de l’industrie pharmaceutique.
La découverte d’un traitement pour la maladie d’Alzheimer a échappé aux scientifiques pendant des décennies. Bien qu’il existe des médicaments pour traiter les symptômes des stades précoce et intermédiaire de la maladie d’Alzheimer, un seul médicament a été approuvé par la Food and Drug Administration (FDA) américaine pour traiter les plaques de protéines associées à la maladie d’Alzheimer : l’aducanumab.
Ce médicament, vendu sous le nom de marque Aduhelm et développé par Biogen, a fait l’objet de vives controverses l’année dernière. Alors qu’on envisageait de l’autoriser en 2021, plusieurs responsables de la FDA ont déclaré qu’il n’y avait pas suffisamment de preuves de ses bienfaits pour justifier une autorisation.
Néanmoins, l’agence a approuvé le médicament, dont le prix de Biogen est de 56 000 dollars. Cela a provoqué la démission de trois responsables de la FDA, dont l’un a déclaré qu’il n’y avait “aucune preuve valable que le médicament fonctionne”.
Le NIH a dépensé environ 1,6 milliard de dollars pour des projets qui mentionnent les amyloïdes au cours de cette année fiscale, soit environ la moitié de son financement global pour la maladie d’Alzheimer. Les scientifiques qui avancent d’autres causes potentielles de la maladie d’Alzheimer, comme un dysfonctionnement immunitaire ou une inflammation, se plaignent d’avoir été mis sur la touche par la “mafia de l’amyloïde”. Selon M. Forsayeth, l’hypothèse amyloïde est devenue “l’équivalent scientifique du modèle ptolémaïque du système solaire”, dans lequel le Soleil et les planètes tournent autour de la Terre.
Les conséquences immédiates et évidentes sont le gaspillage des fonds du NIH et de la recherche scientifique, car les gens utilisent ces résultats comme point de départ pour leurs propres expériences.
Thomas Südhof – Université de Stanford
On ne peut pas tricher pour guérir une maladie. La biologie s’en moque.
Cette vidéo est le cinquième épisode de la série réalisée avec le média Kairos.
Dans le premier épisode, nous avons vu qu’il n’y a eu aucune hécatombe nulle part en Europe, ni en 2020, ni en 2021.
Dans le deuxième épisode nous avons vu qu’il n’y a pas eu la saturation hospitalière annoncée.
Dans le troisième épisode nous avons vu qu’il n’y a pas non plus eu un « déferlement » de malades. Il y a eu bien moins de malades comptabilisés que pendant les épisodes dit « grippaux » du passé. Nous vivons une épidémie de « cas » entretenue par les fameux « tests » qui n’ont pas de rapport avec la moindre maladie.
Dans le quatrième épisode nous avons vu le moteur même de la fraude : l’utilisation de codes spécifiques par les hôpitaux de façon à produire les « bonnes » statistiques.
Dans ce cinquième épisode nous découvrons le socle de l’idéologie sanitaire actuelle qui ne repose sur aucune expérience et est contredite par les statistiques : la contamination.
Via : https://nouveau-monde.ca/le-mythe-de-la-contamination-epidemique/
SHANGHAI, 16 juin (Reuters) – Une manifestation prévue par des centaines de détenteurs de comptes bancaires dans le centre de la Chine en vue d’obtenir l’accès à leurs fonds gelés a été déjouée parce que les autorités ont fait passer au rouge leur pass sanitaire, ont déclaré plusieurs des détenteurs de comptes à Reuters.
Les déposants avaient prévu de se rendre cette semaine dans la province centrale du Henan en provenance de toute la Chine pour protester contre le gel, depuis près de deux mois, d’au moins 178 millions de dollars de dépôts, qui a empêché les entreprises de payer leurs employés et les particuliers d’accéder à leurs économies.
Les groupes de défense des droits ont prévenu que la Chine pourrait utiliser sa vaste infrastructure de surveillance COVID pour étouffer la dissidence. Sans un code vert sur l’application de leur smartphone, les citoyens perdent l’accès aux transports publics et aux espaces tels que les restaurants et les centres commerciaux, ainsi que le droit de voyager à travers le pays.
“Ils nous mettent des menottes numériques”, a déclaré un déposant de la province du Sichuan, prénommé Chen, qui a refusé d’utiliser son nom complet par crainte de représailles du gouvernement.
Le gouvernement provincial du Henan et le ministère de la Sécurité publique n’ont pas répondu aux demandes de commentaires.
La Commission nationale de la santé a déclaré dans une note envoyée à Reuters jeudi que l’utilisation des codes sanitaires ne devait pas être étendue sans autorisation et ne pouvait être attribuée autrement que dans le cadre de la prévention et du contrôle de l’épidémie.
Après les récentes épidémies de COVID, certaines régions de Chine ont demandé aux voyageurs d’enregistrer leurs projets en ligne.
Un homme nommé Liu, qui vit dans la province de Hubei, a constaté que son code sanitaire était devenu rouge le matin du 12 juin après s’être enregistré la veille pour se rendre dans le Henan.
Liu avait prévu de se rendre à une manifestation prévue lundi à Zhengzhou, la capitale provinciale du Henan, où il espérait récupérer son argent. Cette manifestation aurait été la dernière en date des nombreuses manifestations de ce type organisées dans le Henan ces derniers mois.
Plus de 200 déposants ont été bloqués de la même manière lorsque leur code sanitaire est devenu rouge, selon les membres d’un groupe WeChat.
Il n’a pas été possible de déterminer si le changement de code avait pour but de bloquer les manifestants ou pour une autre raison, mais trois déposants ont déclaré à Reuters qu’ils connaissaient des personnes qui s’étaient inscrites pour se rendre dans le Henan, qui n’étaient pas liées aux fonds gelés et dont les codes n’étaient pas devenus rouges.
La Yu Zhou Xin Min Sheng Village Bank, la Shangcai Huimin Country Bank et la Zhecheng Huanghuai Community Bank ont gelé les dépôts le 18 avril, et toutes trois ont indiqué aux clients qu’elles mettaient à jour leurs systèmes internes.
Liu, qui a refusé de donner son nom complet par peur des répercussions du gouvernement, a déclaré que son enfant pourrait ne pas être en mesure d’aller à l’école si son code ne redevient pas rapidement vert.
“Je ne peux rien faire, je ne peux aller nulle part. On vous traite comme si vous étiez un criminel. Cela porte atteinte à mes droits fondamentaux”, a déclaré Liu.
Wang Qiong, qui vit dans la ville centrale de Wuhan, a constaté que son code de santé était devenu rouge après s’être inscrite pour se rendre dans le Henan le 11 juin.
“La police avait les détails de mon identité depuis la dernière fois que je suis allée manifester en avril”, a déclaré Wang, qui dit avoir perdu l’accès à 2,3 millions de yuans (341 550 dollars).
D’autres déposants ont déclaré à Reuters qu’ils avaient pu arriver à Zhengzhou en train et en voiture, mais que leurs codes étaient devenus rouges dès qu’ils avaient scanné les codes sanitaires de la ville.
“La variole du singe” – qui aurait pu le voir venir ? Eh bien, apparemment, l’organisation fondée par Ted Turner en 2001, appelée “Nuclear Threat Initiative” (NTI), l’a vu venir lorsqu’elle a publié un rapport en novembre 2021 intitulé “Strengthening Global Systems to Prevent and Respond to High-Consequence Biological Threats” (Renforcer les systèmes mondiaux de prévention et de réponse aux menaces biologiques à haut risque). Le rapport indique qu’en mars 2021, ils se sont associés à la Conférence sur la sécurité de Munich pour réaliser un scénario d’exercice impliquant une “pandémie mondiale mortelle impliquant une souche inhabituelle du virus de la variole du singe qui est apparue dans la nation fictive de Brinia et s’est répandue dans le monde entier en 18 mois… la pandémie fictive a entraîné plus de trois milliards de cas et 270 millions de décès dans le monde.”
“Renforcer les systèmes mondiaux pour prévenir et répondre aux menaces biologiques de haute gravité
Résultats de l’exercice sur table 2021 mené en partenariat avec la en partenariat avec la Conférence de Munich sur la sécurité”
Étonnamment, le scénario prévoyait l’apparition de l’épidémie de variole du singe à la suite d’un acte de bioterrorisme en mai 2022, là où nous en sommes aujourd’hui. Nous avons traité de l’absurdité du gain de fonction[eng sub] impliquant des virus inexistants dans plusieurs autres vidéos[eng sub], et le Dr Stefan Lanka a également démantelé de tels raisonnements fallacieux. Quoi qu’il en soit, le rapport du NTI suggère que ce qu’il faut faire en cas d’épidémie imaginaire, ce sont “des mesures agressives pour ralentir la transmission du virus en empêchant les rassemblements de masse, en imposant des mesures de distanciation sociale et en mettant en place des obligations de port de masque.” Les pays gagnants dans l’hallucination du NTI ont mis en œuvre “des opérations de dépistage et de recherche des contacts à grande échelle et ont renforcé leurs systèmes de soins de santé”.
“Mais, je n’ai pas de virus Peter.”
Leurs graphiques, qui semblent avoir été produits par la calculatrice de Neil Ferguson, montrent que les pays qui ne se conforment pas à leurs restrictions et à leurs interventions médicales s’en sortiront bien plus mal. Le rapport poursuit en affirmant que “tant le scénario de l’exercice que la réponse de COVID-19 démontrent que les actions précoces des gouvernements nationaux ont des impacts positifs significatifs dans la gestion de l’impact de la maladie.” Quand on parle d'”impacts positifs”, on ne sait pas très bien qui est le bénéficiaire, bien qu’on note que “le marché du vaccin COVID dépassera 150 milliards de dollars en 2021.” Dans l’ensemble, le rapport du NTI se lit comme l’Event 201 sous Ritalin. (L’Event 201 a eu lieu le 18 octobre 2019. Il s’agissait d’un exercice impliquant une ” pandémie de coronavirus “, quelques mois seulement avant que la “pandémie” de COVID-19 ne soit déclarée).
La variole du singe attaque juste au bon moment !
https://www.nti.org/wp-content/uploads/2021/11/NTI_Paper_BIO-TTX_Final.pdf
Comme pour le COVID-19, il semble que d’autres parties aient également attendu avec impatience le marché que représenterait une telle “pandémie”. De même, ces voyants préparaient des vaccins pour aller là où aucun vaccin n’était allé auparavant. En l’occurrence, la société de biotechnologie Bavarian Nordic a obtenu l’approbation de la FDA en 2019 pour commercialiser JYNNEOS, un vaccin contre la variole et la variole du singe. D’autres autorités sanitaires ont également été amorcées pour réagir à une condition auparavant rare qui n’a pas préoccupé leurs nations… jusqu’à présent apparemment. Par exemple, le 20 mai 2022, l’Agence britannique de sécurité sanitaire a publié un document intitulé “Recommandations pour l’utilisation de la vaccination avant et après exposition lors d’un incident lié à la variole du singe.” Comme pour le COVID-19, on commence à avoir l’impression que toutes les routes mènent à nouveau aux vaccins….
Ce n’est qu’une question de temps avant que le vaccin “rare” contre la variole du singe n’arrive dans votre quartier.
Maintenant que le décor est planté, nous pouvons entrer dans la “science” de la variole du singe, en commençant par une description officielle de cette prétendue maladie virale. Selon le CDC, “la variole du singe a été découverte en 1958 lorsque deux épidémies d’une maladie ressemblant à la variole se sont déclarées dans des colonies de singes élevés pour la recherche, d’où le nom de “variole du singe”. Le premier cas humain de variole du singe a été enregistré en 1970 en République démocratique du Congo.” Ils poursuivent en affirmant que, “chez l’homme, les symptômes de la variole du singe sont similaires à ceux de la variole, mais plus légers.” La maladie ressemblerait à la grippe, avec en plus un gonflement des ganglions lymphatiques, puis le développement d’une éruption cutanée, et enfin des lésions qui évoluent de macules en vésicules puis en croûtes.
En ce qui concerne la létalité de la variole du singe, le CDC déclare que “en Afrique, il a été démontré que la variole du singe peut causer la mort d’une personne sur dix qui contracte la maladie”. Ce taux de létalité de 10 % a déjà alimenté le discours de peur et a également été utilisé comme taux de létalité dans le scénario catastrophe du NTI sur la variole du singe. Il convient de noter qu’historiquement, la variole du singe est pratiquement inconnue dans les pays développés et que les rares cas concernent généralement des personnes récemment arrivées d’Afrique.
En effet, l’une des seules “épidémies” de variole du singe enregistrées dans les pays développés a eu lieu aux États-Unis en avril 2003. Des cas ont été déclarés dans 6 États et seraient causés par des rongeurs importés du Ghana au Texas. C’était la première fois que la variole du singe était signalée en dehors de l’Afrique et le CDC a publié en 2006 un document analysant l’incident. Ce document indique que “la propagation du virus de personne à personne se ferait principalement par le biais d’exsudats oropharyngés infectieux”, bien qu’il soit clair que cela n’a jamais été scientifiquement établi. Ils continuent à dire que “le virus aurait été transmis par des animaux africains” – en d’autres termes, c’est une autre histoire d’agent pathogène qui saute d’une espèce à l’autre.
Ils ont rapporté que “les personnes dont la maladie s’est déclarée dans les 21 jours suivant l’exposition au MPXV [virus de la variole du singe] et qui ont présenté de la fièvre (définie comme une température corporelle supérieure à 37,4°C) et une éruption vésiculaire pustuleuse ou une éruption (potentiellement non caractérisée) ainsi que des anticorps IgM anti-virus orthopox ont été classées comme des cas probables d’infection”. Selon notre définition, 37,4°C n’est pas une fièvre, c’est une température corporelle normale et nous pensons que 37,6°C et plus sont considérés comme une fièvre. Nous avons noté dans leur tableau qu’ils utilisaient la classification ≥39,4°C, mais cela semble être une erreur car dans un autre article, que nous aborderons bientôt, il s’agissait à nouveau de 37,4°C. Le second article indique même que la “fièvre” peut être subjective. Il semble donc qu’ils utilisent ces critères peu rigoureux et pathologisent un état normal. En outre, le rapport hebdomadaire du CDC du 11 juillet 2003 indique que sur un total de 71 cas, seuls “deux patients, tous deux des enfants, présentaient une maladie clinique grave ; ces deux patients se sont rétablis”. Les autres patients présentaient divers symptômes respiratoires et gastro-intestinaux.
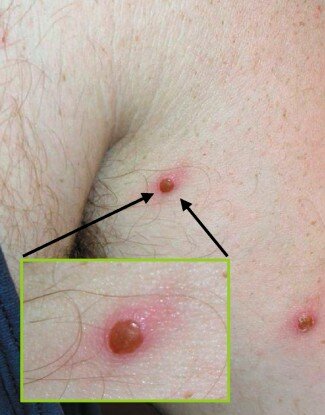
Selon le CDC, les cas ont été confirmés sur la base d’échantillons présentant “l’isolement du virus de la variole du singe, la détection de signatures d’acides nucléiques spécifiques de la variole du singe, des résultats positifs en microscopie électronique ou des résultats positifs en immunohistochimie.” Nous avons examiné les micrographies électroniques présentées par le CDC, notamment l’image ci-dessous d’un échantillon de peau provenant de l’un des patients. La légende nous informe que les particules rondes à droite sont des virions immatures de la variole du singe, tandis que les particules ovales à gauche sont des virus matures. Cependant, il ne s’agit que d’une image statique de tissu mort et aucune conclusion ne peut être tirée quant au rôle biologique des particules imagées. Aucune d’entre elles ne s’est avérée être un parasite intracellulaire pathogène capable de se répliquer et ne devrait donc être appelée “virus”.
En examinant à nouveau le rapport hebdomadaire du CDC de 2003, il apparaît que les 35 “cas confirmés en laboratoire” ont tous fait l’objet de “tests” de réaction en chaîne par polymérase (PCR), et nous avons donc cherché les preuves scientifiques derrière cette affirmation. L’une des citations pour le développement de la détection de la variole du singe par PCR est un article de 2004 intitulé “Real-Time PCR System for Detection of Orthopoxviruses and Simultaneous Identification of Smallpox Virus” (Système PCR en temps réel pour la détection des orthopoxvirus et l’identification simultanée du virus de la variole). Or, un protocole PCR nécessite de connaître les séquences génétiques du prétendu virus de la variole du singe, ce qui nous amène à cet article de 2001 intitulé “Human monkeypox and smallpox viruses : genomic comparison” (Virus de la variole du singe et de la variole humaine : comparaison génomique). Cet article prétendait avoir “isolé” le virus de la variole du singe dans une culture de cellules rénales de singe rhésus provenant d’une croûte d’un patient atteint de la variole du singe. Ici, les virologues reprennent leurs vieux tours en affirmant que : (a) la croûte du patient contient le virus de la variole du singe, et (b) il se trouve maintenant dans leur culture. Ils prétendent avoir séquencé le “génome viral” en se référant à un processus décrit pour le séquençage d’un prétendu virus variolique en 1993.
Mais lorsque nous examinons cet article, aucun virus n’est démontré non plus, simplement une affirmation selon laquelle il a été “isolé” à partir “du matériel d’un patient indien” en 1967. Ils poursuivent en affirmant que “les virions ont été purifiés par centrifugation différentielle et l’ADN viral a été isolé” – cependant, il n’y a aucune démonstration de ce qu’ils ont purifié ou de la façon dont ils ont été déterminés comme étant des virions. Dans aucune de ces expériences, ils n’ont procédé à des contrôles pour voir quelles séquences peuvent être détectées à partir d’autres croûtes d’origine humaine ou de spécimens similaires provenant de personnes malades. C’est ici qu’il faut rappeler aux virologues ce qu’est censé être un virus, c’est-à-dire un parasite intracellulaire capable de se répliquer qui infecte et provoque une maladie chez un hôte. Il ne s’agit pas de détecter des séquences génétiques contenues dans des croûtes et de prétendre qu’elles appartiennent à un virus.
Pour en revenir à l’article du CDC décrivant l'”épidémie” de 2003, il n’est pas clair comment ils ont établi qu’ils pouvaient diagnostiquer la variole du singe en utilisant la PCR. Leur PCR ne peut avoir été calibrée que sur des séquences de provenance indéterminée. En outre, peu importe le type de spécificité analytique de leur protocole PCR, il n’y avait pas de spécificité diagnostique établie – en d’autres termes, il ne s’agissait pas d’un test validé cliniquement, une question qui va au-delà de l’existence ou non du “virus”. (Extrait des directives de la MIQE : La spécificité analytique fait référence au fait que le test qPCR détecte la séquence cible appropriée plutôt que d’autres cibles non spécifiques également présentes dans un échantillon. La spécificité diagnostique est le pourcentage d’individus sans une condition donnée que le test identifie comme négatif pour cette condition).
Les 47 cas américains qu’ils ont fini par décrire ont tous été en contact, d’une manière ou d’une autre, avec des chiens de prairie [rongeurs] africains importés et l’article du CDC conclut que “les individus ont contracté des infections par le MPXV à partir de chiens de prairie infectés ; aucune transmission interhumaine n’a été documentée, mais il y avait de nombreux scénarios potentiels d’infection impliquant des expositions respiratoires et/ou muco-cutanées, des expositions percutanées et/ou par inoculation”. Les auteurs de l’étude ont admis que la conception de l’étude présentait certains problèmes, notamment que “les analyses étaient limitées par la déclaration ou le rappel incomplet des informations par les patients. Et, en raison de la nature rétrospective de l’étude, nous n’avons pas été en mesure d’obtenir des données très détaillées.”
Cependant, même en leur laissant une certaine marge de manœuvre, les incohérences vont encore plus loin. Tout d’abord, personne dans l’incident américain n’est mort de la maladie dont le taux de létalité serait de 10% en Afrique. Il ne fait aucun doute que les taux de létalité incohérents seront attribués à différents “variants”, mais il ne peut y avoir de variants de quelque chose qui n’existe pas.
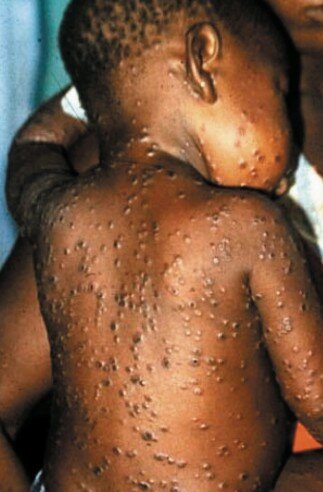
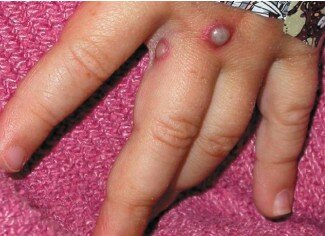
Peu d’images des lésions cutanées signalées lors de l’incident de 2003 étaient disponibles, mais deux des cas américains sont décrits ci-dessous et une image d’un cas de variole du singe en Afrique est présentée à titre de comparaison. Le lecteur peut se faire sa propre opinion, mais ces réactions cutanées ne nous semblent pas du tout comparables.
Enfant africain atteint de la variole du singe
Un enfant américain atteint de la variole du singe
Un homme américain atteint de la variole du singe
Ensuite, le CDC affirme que “le réservoir naturel de la variole du singe reste inconnu. Cependant, les rongeurs africains et les primates non humains (comme les singes) peuvent héberger le virus et infecter des personnes” – en d’autres termes, tout cela est plutôt vague et reste une hypothèse non prouvée. Il est évident que certaines personnes ont été malades aux États-Unis en 2003, mais avec la théorie virale, nous sommes censés croire que le virus est passé de certains chiens de prairie à certains humains et que ces derniers ont été infectés par le prétendu virus… mais alors aucun humain ne pourrait le transmettre à un autre humain. La théorie tombe à plat – un virus doit se propager. Et les schémas historiques des prétendues épidémies de variole du singe n’ont aucun sens – pourquoi le virus est-il transmis à ces personnes si facilement, alors qu’il peut s’écouler une décennie entre les prétendues “épidémies” ?
Malheureusement, l’incident de 2003 a été étudié comme si la théorie de la contagion virale était déjà établie et les autres explications ont été ignorées. Si des personnes sont censées tomber malades à cause de ces rongeurs africains, ne serait-il pas judicieux de vérifier que les animaux ne présentent pas d’autres toxicités, notamment dans leurs excréments, et qu’ils ne sont pas porteurs de tiques ou de parasites ? Nous avons remarqué qu’une autre référence indique que, en ce qui concerne les cas américains, “de nombreuses personnes présentaient des lésions initiales et satellites sur les paumes, les plantes et les extrémités.” Cependant, selon le CDC, la variole du singe commence généralement sur le visage, de sorte que le tableau clinique des cas américains ne correspond pas aux cas généralement décrits en Afrique.
Quoi qu’il en soit, un examen des preuves scientifiques a révélé qu’en ce qui concerne la variole du singe : (a) il n’y a aucune preuve de l’existence d’une particule physique répondant à la définition d’un virus, (b) il n’y a aucune preuve de transmission entre humains et (c) il n’y a aucun moyen de confirmer un diagnostic de variole du singe à moins de croire à des tests cliniquement non validés tels que les kits PCR qui ont été produits. En d’autres termes, si nous assistons à une “pandémie” de variole du singe qui sert de prétexte à l’intensification du terrorisme mondialiste, ce sera à la suite d’une autre pandémie de PCR, et non d’une pandémie naturelle.
Pour ceux d’entre vous qui souhaitent approfondir les problèmes posés par les diverses allégations relatives à la variole du singe, Mike Stone, de ViroLIEgy, a rédigé deux articles intéressants. Le premier article, intitulé “La variole a-t-elle vraiment été éradiquée ?“[FR], traite notamment de l’émergence opportune de la variole du singe alors que la variole était apparemment en voie d’éradication. Le deuxième article s’intitule “Did William Heberden Distinguish Chickenpox From Smallpox in 1767?” (William Heberden a-t-il distingué la varicelle de la variole en 1767 ?). Il souligne le fait que les affections liées à la variole ne sont pas aussi faciles à distinguer les unes des autres que le suggèrent les manuels scolaires et semblent davantage liées à la gravité d’un processus pathologique similaire. Vous pouvez également regarder notre vidéo “Chickenpox Parties and Varicella Zoster Virus?” (Les pox party et le virus varicelle-zona ?) pour voir pourquoi il n’y a pas non plus de preuve de la présence d’un virus dans cette affection connexe.
Du point de vue de la théorie du terrain, c’est une erreur fondamentale d’attribuer la maladie d’une personne à un supposé virus, car les “traitements” qui s’ensuivent ne traitent pas les problèmes sous-jacents. Si quelqu’un ne va pas bien, c’est généralement parce qu’il a une carence en nutriments et qu’il doit rétablir l’équilibre, ou parce qu’il a été exposé à des toxines environnementales et qu’il doit aider son corps à se désintoxiquer. Les guerres contre de prétendus agents pathogènes, qui impliquent de traiter tout le monde de la même manière avec des restrictions des droits civiques et des vaccins, ne relèvent certainement pas de la santé. Il est bon de voir que de plus en plus de personnes prennent conscience de la fraude du COVID-19[FR]. On peut donc espérer qu’une escroquerie à la variole du singe, si elle est tentée, apportera encore plus de lumière à la situation. Comme toujours, votre santé est entre vos mains, et non entre celles d’une secte mondialiste et de ses acolytes.
Article original (anglais) : https://drsambailey.com/viruses/monkeypox-mythology/
Plus de ressources : https://viroliegy.com/2022/05/24/monkey-business/
Le 8 mai 1980, l’Organisation mondiale de la santé (OMS) a annoncé l’éradication complète du “virus” de la variole dans le monde. Ce jour est considéré comme l’une des plus grandes réussites de la médecine moderne. L’humanité s’est unie dans un effort mondial et a finalement vaincu un ennemi mortel en utilisant le “miracle” médical de la vaccination. Cette réussite triomphante a été utilisée pour justifier toutes les campagnes de vaccination depuis lors. La disparition de la variole par le biais d’une injection meurtrière a été fortement mise en avant pour convaincre le public de l’utilité des vaccins expérimentaux à ARNm que l’on nous impose actuellement dans le cadre de la campagne de terreur ” SARS-COV-2 “.
Le 8 mai 1980, la variole a été officiellement déclarée éradiquée au niveau mondial.
Cependant, que se passe-t-il si l’éradication de la variole ne se vérifie pas dans les faits ? Et si les mêmes symptômes de la maladie connue sous le nom de variole existaient encore aujourd’hui ? Et si ces symptômes n’avaient jamais disparu et avaient reçu de nouveaux noms et identités ? Et si les vaccins utilisés pour éradiquer ce “virus” provoquaient en fait les mêmes symptômes que la variole, voire pire ?
Voyons ce que nous pouvons découvrir sur cette prétendue éradication de l’une des “maladies les plus mortelles de tous les temps”.
L’appel de 1958 pour l’éradication mondiale de la variole
En 1958, alors qu’un vaccin contre la variole était utilisé depuis plus d’un siècle et que la prévalence de la maladie avait déjà diminué, l’OMS a décidé qu’il était enfin temps de faire pression pour l’éradication du “virus”. L’objectif était de créer une infrastructure de vaccination dans le monde entier afin d’éradiquer cette maladie une fois pour toutes. Heureusement pour les grands pays comme les États-Unis, la variole avait déjà disparu. Ce sont les petits pays qui ont eu besoin que les États-Unis et l’Union soviétique interviennent et fournissent 150 millions de doses de vaccins pour assurer la victoire.
Vaccin contre la variole : Le bon, la brute et le truand
“Le premier grand effort d’éradication de la variole a été lancé en 1950 dans le but d’éliminer la variole dans les Amériques. En 1958, l’Assemblée mondiale de la santé a adopté une résolution appelant à l’éradication mondiale de la variole. Bien que certains pays aient mis en place des programmes d’éradication de la variole, il n’existait aucune infrastructure coordonnée. De nombreux programmes ont échoué en raison d’un approvisionnement insuffisant en vaccins et de ressources limitées. La forme la plus virulente de la variole, la variole majeure, était répandue aux États-Unis au XIXe siècle, mais seules deux grandes épidémies ont eu lieu entre 1900 et 1925. En revanche, la forme plus bénigne de la variole (variola minor) était courante jusque dans les années 1930. Après 1949, il n’y a plus eu de cas endémiques de variole aux États-Unis, mais la maladie est restée un problème grave dans les pays moins développés. En 1966, la variole restait endémique dans 33 pays. Après un long débat, l’Assemblée mondiale de la santé a approuvé l’octroi de 2,4 millions de dollars pour lancer un programme mondial d’éradication au cours des dix années suivantes. Au début de la campagne, l’Union soviétique et les États-Unis ont fait don de plus de 150 millions de doses de vaccin. À peu près à la même époque, l’aiguille bifurquée a été mise au point, ce qui a simplifié l’administration et réduit le volume de vaccin nécessaire.“
Comme le montre l’OMS en 1958, la vaccination de la population contre la variole fait l’objet d’une pression accrue, en particulier dans les pays les plus pauvres. Ces efforts ont été déployés malgré l’échec des campagnes de vaccination répétées dans certaines régions et malgré les dangers connus liés à l’utilisation du vaccin. L’OMS a demandé une vaccination supplémentaire et la revaccination de certaines populations. En d’autres termes, l’OMS a préconisé une campagne de vaccination de masse avec des rappels tout en demandant des études de sécurité sur les vaccins eux-mêmes. Cela vous rappelle-t-il quelque chose ?
Un vaccin dangereux
Quand le vaccin est plus mortel que la menace d’un “virus éradiqué”.
Alors qu’il semblait que la sécurité des vaccins était encore remise en question en 1958, nous avons la chance, avec le recul, de voir quels types d’effets dévastateurs ont finalement été découverts à long terme.
LES EFFETS INDÉSIRABLES DE LA VACCINATION
Fréquence et caractères cliniques
“Le vaccin antivariolique est moins sûr que les autres vaccins utilisés couramment aujourd’hui. Le vaccin est associé à des effets indésirables connus qui vont de légers à graves. Les réactions légères au vaccin comprennent la formation de lésions satellites, de la fièvre, des douleurs musculaires, une lymphadénopathie régionale, de la fatigue, des maux de tête, des nausées, des éruptions cutanées et une douleur au site de vaccination.13,18,19 Un essai clinique récent a rapporté que plus d’un tiers des personnes vaccinées ont manqué des jours de travail ou d’école en raison de ces symptômes légers liés au vaccin.18”
“En revanche, la plupart des personnes qui reçoivent le vaccin contre la variole ou la variole du singe n’ont que des réactions mineures, comme une légère fièvre, de la fatigue, des glandes enflées, des rougeurs et des démangeaisons à l’endroit où le vaccin est administré. Toutefois, ces vaccins présentent également des risques plus graves.
Sur la base de l’expérience passée, on estime qu’entre 1 et 2 personnes sur 1 million de personnes vaccinées mourront à la suite de complications potentiellement mortelles liées au vaccin.”
Il semblerait que les seuls symptômes des réactions légères au vaccin antivariolique soient la maladie elle-même. À cela s’ajoute la probable sous-estimation par le CDC des décès associés au vaccin.
En 2003, le président Bush a pris la décision de rendre la vaccination contre la variole obligatoire pour tout le personnel militaire et de recommander le vaccin à un demi-million de professionnels de santé. Bien que la variole ait été déclarée éradiquée au niveau mondial 23 ans avant la décision de Bush Jr. et que la maladie elle-même ait été considérée comme non endémique aux États-Unis depuis 1949, la menace que le “virus” éradiqué soit utilisé comme arme biologique a été utilisée pour justifier cette décision. Les reportages de CBS de l’époque dressent un tableau très négatif du vaccin :
” Le vaccin a été créé en 1796. Le vaccin utilisé aujourd’hui est essentiellement le même, dit Mme Offit. “Nous avons tendance à penser que les vaccins sont très sûrs et tous efficaces, ce qu’ils sont. Mais tous les vaccins que nous utilisons aujourd’hui sont le fruit de la technologie moderne. Ce n’est pas le cas du vaccin antivariolique. Il présente un profil d’effets secondaires que nous n’accepterions pas pour les vaccins d’aujourd’hui”, ajoute-t-il.
Alors que l’OMS a lancé un appel mondial à l’éradication de la variole en 1958, les “poxvirus” n’ont apparemment pas reçu le mémo car un autre événement curieux s’est produit cette année-là : la découverte de la variole du singe. Il s’agissait d’une nouvelle maladie qui rappelait étrangement la variole et qui était censée ne toucher que les singes en captivité utilisés à des fins d’expérimentation. Cependant, douze ans après sa découverte et une décennie avant la déclaration de l’éradication de la variole, la variole du singe a décidé de passer de l’animal à l’homme. Bien sûr, le fait que ce “virus” identique à celui de la variole dans tous les domaines ait sauté du navire pour infecter les humains au plus fort de la campagne de vaccination contre la variole n’est qu’une “coïncidence”. Ou se pourrait-il que les mêmes symptômes de maladie associés à la variole aient été renommés, réétiquetés et vendus comme une nouvelle maladie afin de donner l’apparence d’une campagne d’éradication réussie ? Ils l’ont déjà fait avec la varicelle, comme je l’ai expliqué ici :
Il n’est pas difficile de voir que le même tour a été joué ici avec la variole du singe. Deux sources permettent de montrer les étonnantes similitudes entre ces “virus” prétendument distincts. La première provient directement de l’OMS.
Comme le montrent les informations fournies par l’OMS, la variole du singe et la variole sont exactement la même maladie. Elles présentent les mêmes symptômes, le même mode de transmission, le même vaccin et la même réponse théorique des anticorps. La seule différence revendiquée par l’OMS est que la variole du singe est estimée moins mortelle que la variole et qu’elle provient d’un animal de source inconnue alors que la variole ne se trouve que chez l’homme. Ces deux différences sont d’ailleurs théoriques.
Si l’OMS n’était pas suffisamment convaincante quant à la nature identique de ces deux maladies, des extraits du classique de 1988 Smallpox and its Eradication de Frank Fenner, pourraient convaincre. Extrait du chapitre 29 de ce document de près de 1800 pages :
“La variole du singe chez l’homme a été reconnue pour la première fois en 1970 ; il s’agit d’une maladie systémique grave avec une éruption pustulaire généralisée, que l’on ne peut distinguer cliniquement de la variole. Outre les virus de la variole et de la variole du singe, 7 autres espèces de poxvirus, appartenant à 4 genres, peuvent provoquer des lésions chez l’homme (Tableau 29.1). Bien que l’infection par chacun de ces virus produise tout au plus des symptômes légers et généralement seulement une lésion cutanée localisée, les maladies en question ont présenté un problème de diagnostic potentiel lors de l’éradication mondiale de la variole, car les particules virales trouvées dans les lésions par examen au microscope électronique pouvaient être confondues avec celles du virus variolique.”
“La découverte de la variole du singe humaine en Afrique centrale en septembre 1970 a été suivie de la démonstration que 4 cas de variole présumée au Libéria et 1 cas en Sierra Leone en 1970, et 1 cas chacun au Nigeria et en Côte d’Ivoire en 1971 (Foster et al ., 1972) étaient des cas de variole du singe humaine (Lourie et al ., 1972). Une série d’études coordonnées en laboratoire et sur le terrain a été organisée pour déterminer l’incidence de la maladie, étudier ses caractéristiques cliniques et son épidémiologie et rechercher le ou les réservoirs animaux du virus.”
Il est clair que la variole du singe et la variole présentent exactement les mêmes symptômes. On dit que la variole du singe est cliniquement indiscernable de la variole. Il est impossible de les différencier sous microscope électronique car les particules sont exactement les mêmes. Il a été admis que si un réservoir animal de variole était découvert, il ne pourrait pas être éradiqué. Ainsi, au lieu de prétendre que le “virus” de la variole infectait aussi bien les animaux que les humains, ce qui aurait détruit l’histoire de l’éradication, un nouveau “virus” a été créé afin de dire qu’un “virus” identique était passé de l’animal à l’homme. C’est ainsi qu’ils peuvent s’en sortir lorsqu’ils disent que la variole a été éradiquée tout en affirmant que la même maladie existe mais qu’elle est causée par un “virus” différent. Ainsi, les virologues peuvent avoir le beurre et l’argent du beurre.
La lymphadénopathie est-elle spécifique de la variole du singe ?
Les virologues tentent toutefois de créer l’illusion qu’il s’agit de maladies distinctes causées par des “virus” différents en affirmant que la lymphadénopathie est une caractéristique déterminante de la variole du singe. Ils affirment que l’hypertrophie des ganglions lymphatiques est spécifique de la variole du singe et n’a pas été observée dans le cas de la variole. Cependant, cette histoire tombe à l’eau si la lymphadénopathie est également présente dans les cas de variole et ne l’est pas dans tous les cas de monkeypox. Jetons un coup d’œil et voyons si leur fiction tient la route :
Cette première source de novembre 2020 affirme que l’hypertrophie des ganglions lymphatiques n’est pas toujours présente dans les cas de monkeypox :
“Le monkeypox humain ressemble à la variole, avec une éruption cutanée et des signes constitutionnels, mais les symptômes sont généralement plus légers et, contrairement à la variole, les ganglions lymphatiques sont généralement (mais pas toujours) hypertrophiés. Le plus souvent, la maladie commence par des symptômes non spécifiques, semblables à ceux de la grippe, qui peuvent comprendre un malaise, de la fièvre, des frissons, des maux de tête, des maux de gorge, des myalgies, des maux de dos, de la fatigue, des nausées, des vomissements et une toux non productive. La lymphadénopathie peut être régionale ou généralisée et touche le plus souvent les ganglions lymphatiques submandibulaires, postauriculaires, cervicaux et/ou inguinaux.”
Alors que cette deuxième source de 2018 indique que le gonflement des ganglions lymphatiques n’est pas habituellement observé avec la variole, nous pouvons donc en déduire qu’il existe des cas où le gonflement des ganglions lymphatiques s’est produit :
Quels sont les symptômes de la variole du singe ?
“Chez l’homme, les signes et les symptômes de la variole du singe sont similaires à ceux de la variole, mais ils sont généralement plus légers. La variole du singe provoque de la fièvre, des maux de tête, des maux de dos, un gonflement des ganglions lymphatiques (qui n’est généralement pas observé dans le cas de la variole), des maux de gorge et de la toux.”
Les virologues avaient besoin d’un nouveau symptôme spécifique pour faire accepter l’idée que la variole du singe est en quelque sorte différente de la variole. Cependant, il semblerait, d’après ces sources, que le gonflement des ganglions lymphatiques ne soit pas toujours un symptôme de la variole et qu’il puisse également accompagner la variole. Ainsi, ce symptôme ne peut guère être considéré comme spécifique de la variole du singe ni comme un moyen de différencier les deux.
Comment peut-on alors prétendre que le gonflement des ganglions lymphatiques n’est pas une caractéristique de la variole ? S’agit-il vraiment d’un symptôme qui n’a pas été retrouvé dans les cas de la maladie ou se peut-il que l’adénopathie n’ait pas été recherchée lors de l’examen ? Une troisième source datant de 2012 soutient cette dernière hypothèse en affirmant que le gonflement des ganglions n’était pas bien décrit pour la variole, car très peu d’attention était portée à ce symptôme lors de l’examen. Cependant, une hypertrophie (agrandissement) et une hyperamélie (excès de sang) des glandes lymphatiques ont été notées dans les cas de variole. On disait que cette hypertrophie était due à une rétention d’eau. Il est également affirmé dans cette source que les cas de variole du singe ont très probablement été diagnostiqués comme des cas de variole (ou logiquement l’inverse) et que la variole du singe n’a même pas été reconnue comme une maladie distincte avant 1970, ce qui signifie qu’il s’agissait jusqu’alors de la même maladie :
“La pathologie des ganglions lymphatiques dans les cas de variole naturelle est mal décrite. Councilman et al. (1904) notent que dans la littérature antérieure au 20ème siècle, ‘très peu d’attention a été accordée à l’état des ganglions lymphatiques dans la variole’. Dans son étude de cas, Bras (1952) rapporte que les ganglions lymphatiques n’étaient pas examinés régulièrement et que leur description se limite à trois phrases. D’après les données disponibles, les modifications ganglionnaires brutes les plus fréquemment rapportées sont l’hypertrophie et l’hyperémie ; cependant, dans de nombreux cas, les ganglions lymphatiques sont apparemment normaux. Sur le plan histologique, l’hypertrophie, si elle est présente, semble être due principalement à un œdème et à une congestion. Councilman et al. (1904) déclarent spécifiquement que “l’élargissement du ganglion est plus dû à l’œdème qu’à l’hyperplasie cellulaire”. Une histiocytose sinusale et une hémorragie multifocale avec érythrophagocytose et fibrine abondante sont également rapportées. Comme pour la rate, de nombreux auteurs décrivent également de multiples foyers de nécrose et de lymphocytolyse, avec ou sans bactéries ; cependant, une association avec un type de maladie spécifique n’est pas toujours faite. Dans quelques rapports, la nécrose avec des bactéries intralésionnelles serait plus fréquente avec la maladie hémorragique.”
“Avant l’éradication de la variole, les infections humaines à MPXV étaient probablement diagnostiquées à tort comme des infections à VARV en raison de la prévalence de la variole et de la similitude de la présentation et de l’évolution de la maladie cutanée. Le monkeypox n’a pas été reconnu comme une maladie distincte de la variole jusqu’en 1970, lorsque l’élimination de la variole en République démocratique du Congo a révélé la persistance d’une maladie semblable à la variole (Fenner et al., 1988b).”
Il est clair que la lymphadénopathie n’est pas spécifique à la variole du singe, mais il peut y avoir une raison à l’augmentation de ce symptôme si c’est vraiment le cas. Dans la section précédente sur les effets secondaires de la vaccination antivariolique, la lymphadénopathie et le gonflement des glandes ont été mis en évidence comme des réactions connues à la vaccination. Ceci a été documenté par une étude sur les effets indésirables datant de 1968 :
“Le lymphandénite postvaccinal est l’expression désignant les modifications réactives qui se produisent dans les ganglions lymphatiques en réponse à une vaccination antivariolique.”
“De 1983 à 1991, 4649 doses de vaccin antivariolique ont été administrées, dont 57% en 1989-91. La proportion de primo-vaccinations est passée de 4% en 1983-88 à 14% en 1989-91. Parmi les personnes vaccinées, 93% n’ont signalé aucun signe ou symptôme après la vaccination. Les effets indésirables signalés étaient légers : lymphadénopathie, fièvre ou frissons, et sensibilité au site de vaccination. Aucun effet indésirable grave n’a été signalé. Cependant, une personne vaccinée a signalé un avortement spontané 5 mois après la primovaccination (16).”
Même le CDC connaissait cette réaction à la vaccination antivariolique et la considérait comme normale :
Les réactions normales qui ne nécessitent pas de traitement spécifique comprennent la fatigue, les céphalées, les myalgies, les lymphadénopathies régionales, la lymphangite, le prurit et l’œdème au site d’inoculation, ainsi que les lésions satellites, qui sont des lésions secondaires bénignes, proximales aux lésions centrales de la vaccination.
https://www.aafp.org/afp/2003/0415/p1827.html
Si l’on s’en tient aux faits, le vaccin antivariolique était connu pour provoquer une lymphadénopathie ainsi que tous les autres symptômes associés à la variole du singe et à la variole. Une campagne de vaccination de masse a été lancée dans les années 1950 et l’OMS a appelé à l’éradication mondiale de la variole en 1958. Par coïncidence, la variole du singe a également été découverte en 1958 chez des singes captifs utilisés pour des expériences de vaccination contre la polio. En 1970, un garçon du Zaïre, une région réputée exempte de variole depuis 1968, aurait été le premier cas humain de variole du singe, une maladie que l’on ne peut distinguer cliniquement de la variole, à l’exception du symptôme de lymphadénopathie, une réaction connue à la vaccination antivariolique et un symptôme négligé de la variole. Si l’on examine cette situation de manière critique et logique, il est facile de voir que l’apparition soudaine de la variole était la couverture parfaite pour l’OMS afin d’entretenir le mythe de l’éradication de la variole et de dissimuler les réactions indésirables à la vaccination.
En résumé :
Le premier grand effort d’éradication de la variole a été lancé en 1950 dans le but d’éliminer la variole dans les Amériques.
En 1958, l’Assemblée mondiale de la santé a adopté une résolution appelant à l’éradication mondiale de la variole.
Après 1949, il n’y a plus de cas endémiques de variole aux États-Unis, mais la maladie reste un problème grave dans les pays moins développés.
Au début de la campagne, l’Union soviétique et les États-Unis ont fait don de plus de 150 millions de doses de vaccin.
À peu près à la même époque, l’aiguille bifurquée a été mise au point, ce qui a simplifié l’administration et réduit le volume de vaccin nécessaire.
Dans ses notes de 1958, l’OMS admet le fait que la variole persiste dans certaines régions malgré les campagnes de vaccination répétées
Si l’OMS pousse à l’augmentation de la production de vaccins, elle demande également que soient étudiées les mesures à prendre pour éviter les complications qui pourraient résulter de la vaccination antivariolique.
Ils ont demandé à tous les gouvernements de vacciner, en 1959-1960, la population des pays dans lesquels existent les principaux foyers endémiques de variole.
Ils ont également déclaré qu’en 1961-1962, une vaccination supplémentaire de la population devrait être effectuée dans les foyers où la maladie persiste et que, par la suite, des revaccinations seraient effectuées dans la mesure où cela s’avérerait nécessaire, conformément à l’expérience acquise dans chaque pays.
Enfin, il a été demandé aux médecins et aux institutions scientifiques actives dans le domaine de la microbiologie et de l’épidémiologie de stimuler leurs efforts en vue d’améliorer la qualité et la technologie de la production d’un vaccin antivariolique satisfaisant et résistant à l’influence de la température.
En d’autres termes, l’OMS a préconisé une campagne de vaccination de masse avec des rappels tout en demandant des études de sécurité sur les vaccins eux-mêmes.
Le vaccin antivariolique est “moins sûr” que les autres vaccins couramment utilisés aujourd’hui.
Les réactions légères au vaccin comprennent : la formation de lésions satellites, fièvre, douleurs musculaires, lymphadénopathie régionale, fatigue, maux de tête, nausées, éruptions cutanées, douleur au site de vaccination.
Selon les estimations toujours “précises” du CDC, on estime qu’entre 1 et 2 personnes sur 1 million de vaccinés mourront à la suite de complications potentiellement mortelles liées au vaccin.
Le vaccin antivariolique est mortel et les scientifiques le qualifient de vaccin le plus dangereux connu de l’homme.
En 2003, l’administration Bush a rendu obligatoire la vaccination de tout le personnel militaire contre la variole et a recommandé aux professionnels de santé de recevoir le vaccin.
Cette mesure était motivée par la menace d’une utilisation d’un “virus” éradiqué comme arme biologique.
Le dilemme était décrit comme suit :
Ne pas vacciner la population contre la variole et laisser des millions de personnes vulnérables à l’un des pires fléaux connus de l’homme.
Ou traiter les gens avec un vaccin qui est extrêmement efficace pour bloquer la maladie mais qui peut provoquer des réactions dangereuses, parfois mortelles.
Le vaccin a été créé en 1796 et le vaccin utilisé aujourd’hui est essentiellement le même.
Le profil des effets secondaires du vaccin antivariolique ne serait pas accepté pour les vaccins actuels.
Le vaccin antivariolique est fabriqué à partir d’un cousin biologique faible du “virus” de la variole.
En 1958, le “virus” de la variole du singe a été découvert chez des primates en cage utilisés pour des expériences scientifiques et a fini par être transmis à l’homme en 1970.
Selon l’OMS, la présentation clinique de la variole du singe ressemble à celle de la variole.
La variole du singe est une zoonose “virale” (un “virus” transmis à l’homme par les animaux) dont les symptômes sont similaires à ceux observés dans le passé chez les patients atteints de variole.
La variole du singe a été identifiée pour la première fois chez l’homme en 1970, en République démocratique du Congo (alors appelée Zaïre), chez un garçon de 9 ans, dans une région où la variole avait été éliminée (comme par hasard) en 1968.
Malgré son nom, le réservoir naturel de la variole du singe n’a pas encore été identifié, mais les rongeurs seraient le plus probable.
Le diagnostic différentiel clinique à prendre en compte comprend d’autres maladies à éruptions cutanées, telles que la varicelle, la rougeole, les infections cutanées bactériennes, la gale, la syphilis et les allergies liées aux médicaments.
La lymphadénopathie (nous y reviendrons plus tard) pendant la phase prodromique de la maladie peut être une caractéristique clinique permettant de distinguer la variole du singe de la varicelle ou de la variole.
La réaction en chaîne par polymérase (PCR) est le test de laboratoire privilégié en raison de sa précision et de sa sensibilité.
Pour cela, les échantillons optimaux pour le diagnostic de la variole du singe proviennent des lésions cutanées – le toit ou le liquide des vésicules et des pustules, et les croûtes sèches.
Les tests sanguins PCR ne sont généralement pas concluants en raison de la courte durée de la virémie par rapport au moment du prélèvement de l’échantillon après le début des symptômes et ne doivent pas être prélevés systématiquement sur les patients.
En d’autres termes, le “virus” est en quelque sorte présent dans les lésions cutanées mais pas dans le sang…
Les “orthopoxvirus” ayant une réactivité sérologique croisée, les méthodes de détection des antigènes et des anticorps ne permettent pas de confirmer la spécificité du virus de la variole du singe (c’est-à-dire qu’elles donneraient un résultat positif pour la variole ou tout autre “poxvirus”) et ne sont pas recommandées.
En outre, une vaccination récente ou lointaine avec le vaccin contre la variole (par exemple, toute personne vaccinée avant “l’éradication” de la variole, ou vaccinée plus récemment en raison d’un risque plus élevé) peut entraîner des résultats faussement positifs.
En d’autres termes, les anticorps sont une mesure inutile puisqu’ils indiquent qu’une personne est positive à la variole.
L’OMS admet à nouveau que la présentation clinique de la variole du singe ressemble à celle de la variole, une infection orthopoxvirale apparentée qui a été éradiquée dans le monde entier.
Elle déclare également que, bien que la variole n’existe plus à l’état naturel, le secteur mondial de la santé reste vigilant au cas où elle pourrait réapparaître par des mécanismes naturels, un accident de laboratoire ou une dissémination délibérée.
Dans l’ouvrage Smallpox and its Eradication (1988), il est indiqué que la variole du singe est une maladie systémique grave accompagnée d’une éruption pustuleuse généralisée et qu’elle est cliniquement impossible à distinguer de la variole.
Outre les “virus” de la variole et de la variole du singe, 7 autres espèces de “poxvirus”, appartenant à 4 genres, peuvent provoquer des lésions chez l’homme.
La variole du singe a posé un problème de diagnostic potentiel lors de l’éradication mondiale de la variole, car les particules de “virus” trouvées dans les lésions par examen au microscope électronique pouvaient être confondues avec celles du “virus” de la variole.
En d’autres termes, on a trouvé exactement les mêmes particules non purifiées/non isolées dans les cultures, mais on a prétendu qu’il s’agissait de “virus” différents.
Il était évident que si un réservoir animal du “virus” de la variole existait, l’éradication de la variole serait impossible (et voilà qu’un réservoir animal existe…).
Après la découverte de la variole du singe en 1958, l’OMS a enquêté sur d’autres épidémies.
Les enquêtes qui ont suivi ont révélé 4 autres épidémies signalées et 4 épidémies jusqu’alors non signalées chez les primates, mais aucun cas d’infection chez l’homme.
Dans l’un de ces cas, le “virus” de la variole du singe avait été récupéré dans des cultures de cellules normales de rein de cynomolgus.
Après la découverte de la variole du singe en Afrique en 1970, des sérums ont été collectés chez des singes et d’autres animaux au Zaïre et dans plusieurs pays d’Afrique occidentale.
Des “anticorps spécifiques du virus de la variole du singe” ont été mis en évidence dans les sérums de 8 espèces de singes et de 2 espèces d’écureuils (ce qui va à l’encontre des informations plus récentes de l’OMS selon lesquelles il n’existe pas d’anticorps spécifiques pour la variole du singe)
Bien que des primates d’Asie, d’Afrique et d’Amérique du Sud (et un fourmilier de cette dernière région) aient été infectés par le “virus” de la variole du singe en captivité, rien ne prouve que ce “virus” soit présent naturellement ailleurs qu’en Afrique.
Au cours de la période 1958-1968, un grand nombre de primates ont été importés d’Asie en Europe et en Amérique du Nord, et un plus petit nombre d’Afrique occidentale, principalement pour la fabrication et les tests de sécurité des vaccins contre la poliomyélite.
En d’autres termes, les animaux utilisés pour les tests des vaccins expérimentaux sont tombés malades entre les expériences, pendant le transport dans des conditions horribles
Il a été signalé que des variants appelés “virus de la variole”, qui ressemblaient au “virus” de la variole par tous les tests biologiques, pouvaient être récupérés à partir de certains stocks de laboratoire du “virus” de la variole du singe, soit par passage dans des hamsters, soit par inoculation sur la membrane chorio-allantoïque.
Cette découverte a soulevé d’importantes questions quant à la possibilité d’un réservoir animal du “virus” de la variole, mais ces questions ont été écartées par la suite (rappelez-vous qu’ils ont admis que la variole ne pourrait pas être éradiquée si un réservoir animal était découvert).
Vers 1982, l’accumulation des preuves a convaincu la plupart des laborantins que les “virus de la variole” étaient en fait des souches du “virus” de la variole introduites par inadvertance comme contaminants de laboratoire (comme c’est pratique…).
L'”isolement” de “virus” à partir d’animaux capturés sur le terrain est probablement un événement rare dans les infections à “orthopoxvirus”, dans lesquelles une infection persistante ne se produit pas, et en fait, un seul “isolement” de ce type a été effectué.
La dernière épidémie connue de variole dans la zone de Basankusu s’est produite en 1968 et a comporté 70 cas dont 18 décès.
Plusieurs cas suspects de variole ont été traités à l’hôpital en 1969, mais aucun n’a été confirmé.
Deux cas suspects ont été signalés en 1970 ; l’un d’entre eux s’est avéré être la varicelle, et l’autre a été le premier cas de monkeypox humain à être détecté.
Le premier cas de variole du singe présentait, le 9e jour, une éruption cutanée dont la distribution centrifuge était caractéristique de la variole.
Le patient s’est rétabli et était sur le point de sortir, mais le 23 octobre, il a développé une rougeole (contractée pendant son séjour à l’hôpital) et est décédé 6 jours plus tard (la rougeole était aussi régulièrement confondue avec la variole).
La découverte de cas de variole humaine en Afrique centrale en septembre 1970 a été suivie de la démonstration que 4 cas de variole suspectés au Liberia et 1 cas en Sierra Leone en 1970, et 1 cas au Nigeria et en Côte d’Ivoire en 1971 étaient tous des cas de monkeypox humaine (on ne peut pas avoir des cas de variole qui apparaissent alors qu’elle est “éradiquée…”).
Les virologues qui s’intéressent aux “poxvirus” savaient depuis 1959 que le “virus” de la variole du singe pouvait provoquer une maladie généralisée ressemblant à la variole chez les singes cynomolgus, et dans les années 1960, des cas similaires ont été reconnus chez d’autres espèces de singes et chez les singes anthropoïdes.
Lors de la première réunion du Groupe informel de l’OMS sur la variole du singe et les virus apparentés, qui s’est tenue à Moscou en mars 1969, les experts ont convenu que la première indication que le “virus” récupéré d’une lésion cutanée pourrait être le “virus” de la variole du singe serait l’aspect hémorragique des boutons produits sur la membrane chorioallantoïque après 3 jours d’incubation à 35° C.
Le 23 septembre 1970, les docteurs S. S. Marennikova, E. M. Shelukhina et N. N. Maltseva, du centre collaborateur de l’OMS à Moscou, ont récupéré un “virus” sur la membrane chorio-allantoïque à partir de matériel envoyé par un patient au Zaïre.
Après une incubation de deux jours, les taches étaient “parfaitement typiques” du “virus” de la variole.
Cependant, après un autre jour d’incubation à 35°C, une hémorragie a été observée autour des pustules, une caractéristique jamais observée avec le “virus” variolique et caractéristique du “virus” de la variole du singe.
En d’autres termes, ils ont déterminé que l’apparition hémorragique de la membrane chorioallantoïque après un jour supplémentaire d’incubation était la variole du singe.
Entre-temps, un diagnostic de “virus” de la variole a été établi au centre de collaboration de l’OMS à Atlanta à partir de matériel provenant de deux cas de maladie ressemblant à la variole découverts dans différentes régions du Liberia à la mi-septembre.
Ce diagnostic a suscité une grande inquiétude, car on pensait que le Liberia était exempt de variole depuis 1969.
Ils ont décidé que les isolats devaient être soigneusement examinés au moyen de tests appropriés afin de déterminer s’il pouvait s’agir du “virus” de la variole du singe.
Les isolats libériens, ainsi que les isolats ultérieurs de la Sierra Leone et du Nigeria, se sont avérés avoir les caractéristiques du “virus” de la variole du singe (la magie de la virologie…).
Des dispositions ont été prises pour un examen plus approfondi des isolats du Zaïre et du Libéria et les travaux sur ces isolats ont constitué le principal sujet de discussion lors de la deuxième réunion du Groupe informel sur la variole du singe et les virus apparentés en février 1971.
Les “experts” présents à cette réunion ont convenu que ces isolats étaient bien des “virus” de la variole du singe.
Cette conclusion a été une source de soulagement considérable, car elle excluait la possibilité que la variole se soit reproduite dans les situations épidémiologiques les plus improbables (whew…)
Cliniquement, la variole humaine ressemble beaucoup à une variole discrète de type ordinaire ou, parfois, de type modifié.
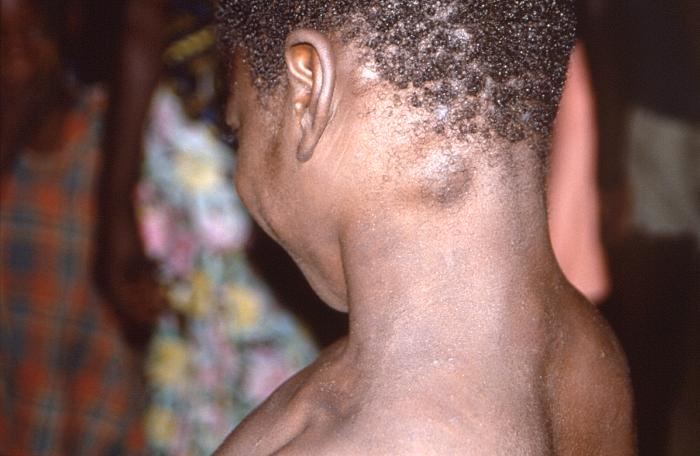
La caractéristique clinique évidente qui différencie la variole du singe de la variole est l’élargissement prononcé des ganglions lymphatiques observé dans la plupart des cas de variole du singe (mais pas dans tous).
L’hypertrophie des ganglions lymphatiques a été observée dans 90 % des 98 cas dans lesquels sa présence ou son absence a été enregistrée et était un signe présent, précédant l’éruption, dans 65 % de ces cas.
L’éruption commence après une maladie prodromique qui dure 1-3 jours, avec de la fièvre, une prostration et généralement une hypertrophie des ganglions lymphatiques.
Comme dans le cas de la variole, les lésions se développent plus ou moins simultanément et évoluent à la même vitesse, à travers des papules, des vésicules et des pustules, avant de s’ombiliquer, de sécher et de desquamer.
Comme dans le cas de la variole, des cicatrices en forme de piqûres peuvent se développer, le plus souvent sur le visage, mais elles ont tendance à diminuer en importance avec le temps.
Tout au long des enquêtes, une grande importance a été accordée à la confirmation en laboratoire des diagnostics clinico-épidémiologiques, d’abord en raison de la présence possible de la variole, puis de la suspicion d’une infection humaine par le “virus de la variole blanche”.
Les méthodes de diagnostic de laboratoire étaient les mêmes que celles utilisées pour la variole, complétées par une sérologie (non spécifique) dans les cas où l’isolement “viral” n’était pas possible.
Cette combinaison a permis de poser des diagnostics positifs dans la grande majorité des cas (qu’en est-il de ceux où le diagnostic n’a pu être posé… ?)
Pratiquement tous les cas trouvés positifs par microscopie électronique l’étaient aussi par culture, et vice versa (pourquoi ne serait-ce pas le cas puisque le matériel de microscopie électronique provient de la culture… ?) mais 60 (22%) des cas ont été vus trop tard pour obtenir du matériel lésionnel et n’ont pu être confirmés que par sérologie (qui, encore une fois, n’est pas spécifique en raison des réactions croisées avec la variole et d’autres “virus”).
L’OMS a entrepris des tests sérologiques à partir de quelque 200 sérums provenant de zones éloignées de ce qui est maintenant reconnu comme la zone d’enzootie de la variole du singe et tous étaient pratiquement négatifs, alors que les sérums de singes du Zaïre prélevés en 1971 et 1973 se sont révélés 14 sur 81 positifs par le test HI et 11 sur 65 par le test de neutralisation
Par la suite, une autre collecte de sérums au Zaïre a donné 24 sérums de singe HI-positifs sur 117 testés et 26 sérums de rongeur HI-positifs sur 245 testés.
Des tentatives ont été faites pour isoler le “virus” sur la membrane chorio-allantoïque des reins de primates, de rats et d’écureuils collectés au Zaïre.
Aucun n’a donné de “virus” de la variole du singe, mais le “virus” de la variole blanche aurait été obtenu à partir de 4 spécimens et le “virus” de la vaccine à partir d’un spécimen.
Dans une autre étude sérologique de l’OMS, 1331 sérums provenant de 45 espèces d’animaux sauvages ont été testés par le test HI comme test de dépistage des anticorps contre les “orthopoxvirus” ; 227 sérums (17%), provenant d’un large éventail d’animaux, ont donné des résultats positifs.
Les 50 sérums provenant de Rattus spp. étaient tous négatifs.
L’analyse ultérieure de certains sérums par des tests d’adsorption radio-immunologique a jeté un doute sur la signification des résultats positifs obtenus par le test IH, puisque aucun des 25 sérums IH-positifs de l’écureuil Heliosciurus rufobrachium n’a donné de résultats positifs par radio-immunologie.
Les reins et les rates de 930 animaux de l’étude de 1979 sur le Zaïre, y compris tous les singes, ont été mis en culture dans des cellules Vero, et le matériel de singe a également été testé sur la membrane chorio-allantoïque, avec des résultats négatifs.
Ce que tout cela signifie, c’est que les résultats des anticorps ne veulent absolument rien dire.
Le chapitre conclut que la variole du singe s’est avérée en 1970 être l’agent causal d’une infection humaine généralisée qui ressemblait cliniquement à la variole.
On a prétendu que la lymphadénopathie était une caractéristique essentielle de la variole du singe, mais elle n’est pas toujours présente.
Le plus souvent, la maladie commence par des symptômes non spécifiques, semblables à ceux de la grippe.
La pathologie des ganglions lymphatiques dans les cas de variole naturelle est mal décrite.
Councilman et al. (1904) notent que dans la littérature antérieure au 20e siècle, “très peu d’attention a été accordée à l’état des ganglions lymphatiques dans la variole”.
Dans sa série de cas, Bras (1952) rapporte que les ganglions lymphatiques n’étaient pas examinés régulièrement et que leur description se limite à trois phrases.
Parmi les données disponibles, les modifications ganglionnaires brutes les plus fréquemment rapportées sont l’hypertrophie et l’hyperémie
Histologiquement, l’hypertrophie, si elle est présente, semble être due principalement à l’œdème et à la congestion.
Councilman a spécifiquement déclaré que “l’élargissement du ganglion est davantage dû à l’œdème qu’à l’hyperplasie cellulaire”.
Avant l’éradication de la variole, les infections humaines à MPXV étaient probablement diagnostiquées à tort comme des infections à VARV en raison de la prévalence de la variole et de la similitude de la présentation et de l’évolution de la maladie cutanée.
La variole du singe n’a pas été reconnue comme une maladie distincte de la variole jusqu’en 1970, lorsque l’élimination de la variole en République démocratique du Congo a révélé la persistance d’une maladie semblable à la variole.
La lymphadénopathie et le gonflement des ganglions lymphatiques sont des réactions connues à la vaccination antivariolique.
La lymphandénite postvaccinale est l’expression désignant les changements réactifs qui se produisent dans les ganglions lymphatiques en réponse à une vaccination antivariolique.
Les réactions indésirables signalées lors de la vaccination antivariolique dans les années 1980 comprenaient une lymphadénopathie, de la fièvre ou des frissons et une sensibilité au niveau du site de vaccination.
La variole ou… ?
La variole a-t-elle réellement été éradiquée comme le prétendait l’OMS en 1980 ? Cela dépend de la définition du terme “éradiquer”. Si l’on se réfère à l’élimination du nom “variole” utilisé pour décrire un ensemble de symptômes reconnaissables de la maladie, alors la réponse est un OUI absolu puisque le nom a été retiré et remplacé par celui de variole du singe (ou de varicelle, de rougeole, de rubéole, etc.). Si l’on se réfère à la suppression complète des symptômes associés au nom, alors la réponse est un NON catégorique, car les mêmes symptômes de la maladie apparaissent dans diverses maladies sous des noms différents et sont même acquis par des vaccinations de routine. L'”éradication” de la variole n’était rien de plus qu’un écran de fumée utilisé pour vendre au monde le “miracle” de la vaccination. C’est une affirmation qui ne résiste pas à un examen approfondi.
Source (anglais) : https://viroliegy.com/2022/01/05/was-smallpox-really-eradicated/
Plus de ressources : http://whale.to/vaccines/smallpox.html
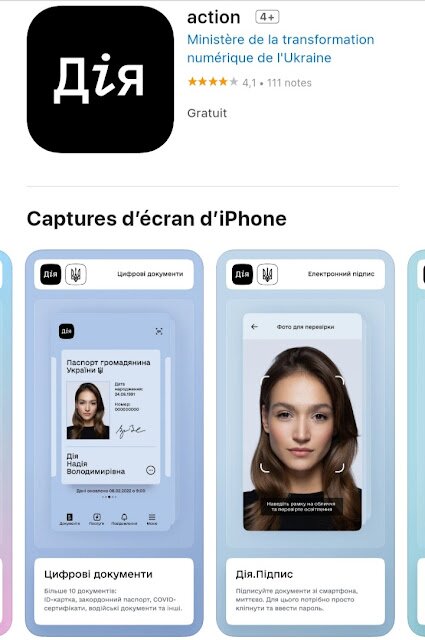
Vers 1′:50″, un représentant du gouvernement ukrainien annonce que les citoyens et entreprises qui ont perdu leur travail, leurs économies et leurs affairses, pourront bénéficier d’une aide gouvernementale.
Le ou les versements en argent seront faits à partir d’une application du “Ministère de la transformation numérique de l’Ukraine”, dont le nom de l’application semble se traduire par “Action”. Les citoyens devront la télécharger, s’inscrire et suivre les instructions afin de recevoir cette “compensation de guerre”. La vaccination est aussi obligatoire pour recevoir la ou les sommes.
Les détails donnés sur l’application “Action” nous indiquent clairement qu’il s’agit d’un système d’IDENTIFICATION NUMÉRIQUE…
On peut lire, dans la description de l’application :
“Après avoir installé “l’action”, vous pourrez :
– utiliser des documents numériques ;
– recevoir des services publics en quelques clics ;
– partager des copies de documents numériques.
Après avoir installé l’action, vous pourrez :
– utiliser des documents numériques ;
– recevoir des services publics en quelques clics ;
– partager des copies de documents numériques.
Les originaux en papier et en plastique peuvent désormais être laissés à la maison. Pour obtenir des versions numériques de vos documents, il vous suffit de télécharger l’action et de vous connecter. Ils apparaîtront automatiquement dans l’application si vos données sont dans les registres.
Documents numériques en action :
– Passeport d’un citoyen ukrainien sous la forme d’une carte d’identité.
– Passeport biométrique.
– Carte de contribuable (RNOKPP).
– Le permis de conduire.
– Certificat d’immatriculation du véhicule.
– Police d’assurance du véhicule.
– Carte d’étudiant.
– Certificat de réinstallation (IDP).
– L’acte de naissance de votre enfant.
– Certificats COVID-19.
Services publics en ligne :
– Paiement des amendes pour infractions au code de la route.
– Paiement de dettes dans le cadre d’une procédure d’exécution.
– Paiement de l’impôt sur le revenu des personnes physiques.
– Soumission des déclarations FOP.
– Remplacement du permis de conduire.
– Enregistrement de résidence.
– Paiement des prestations administratives par QR-code.
– Assistance ponctuelle aux propriétaires uniques et aux salariés.”
Nick Giambruno : Doug, vous êtes une sommité mondiale en matière d’investissement en période de crise. Parlez-nous un peu de votre parcours dans ce domaine.
Doug Casey : Après la sortie de mon deuxième livre, Crisis Investing, en 1979, j’ai commencé à publier une newsletter du même nom. J’ai utilisé le symbole chinois de la crise comme logo. Il s’agit en fait d’une combinaison de deux symboles : celui du danger et celui de l’opportunité. Le danger est ce que tout le monde voit ; l’opportunité n’est jamais aussi évidente que le danger, mais elle est toujours là.
Spéculer sur les marchés en crise est le moyen ultime d’être anticonformiste, c’est-à-dire d’acheter quand personne d’autre ne veut acheter.
Il est vrai, en règle générale, que vous voulez “profiter de la tendance”. Mais il arrive toujours un point d’inflexion où les tendances changent parce qu’un marché devient soit fortement surévalué, soit fortement sous-évalué. Et lorsqu’un marché est en baisse de 90 % ou plus, vous devez, par réflexe, l’examiner, quelles que soient les mauvaises nouvelles, et voir si c’est un secteur où vous souhaitez placer des capitaux spéculatifs.
Nick Giambruno : Des fortunes colossales ont été constituées au cours de l’histoire grâce à des investissements de crise. Le baron Rothschild avait-il raison de dire que le moment d’acheter est celui où le sang coule dans les rues ?
Doug Casey : C’est un aphorisme très célèbre, bien sûr. Il est censé avoir été inspiré par la bataille de Waterloo, lorsqu’il a acheté des titres britanniques alors que la situation était incertaine.
Il a pu réussir ce coup parce qu’il s’est assuré d’obtenir l’information sur la victoire de Wellington sur Napoléon un jour avant tout le monde. Il a reconnu que l’Europe traversait une période de crise majeure.
Nick Giambruno : Cela me fait penser aux oligarques russes, qui sont devenus oligarques en premier lieu parce qu’ils ont fait des investissements de crise, c’est-à-dire qu’ils ont acheté lorsque le sang coulait dans les rues et ont récupéré certains des joyaux de l’économie russe pour quelques centimes.
Doug Casey : C’est intéressant avec les oligarques, car en Union soviétique, tout le monde recevait des certificats, qui étaient échangés contre des actions d’entreprises en cours de privatisation. La personne moyenne n’avait aucune idée de ce qu’ils étaient ou de comment les évaluer. Les personnes qui sont devenues des oligarques ont pu les acheter pour quelques centimes, en profitant de l’hystérie publique négative qui a suivi l’effondrement de l’Union soviétique.
C’est donc un thème récurrent – acheter quand le sang coule dans les rues. C’est l’essence même de la spéculation : profiter des distorsions du marché d’origine politique, ou profiter des aberrations de la psychologie de masse.
Je veux dire, tout le monde connaît la vieille expression “acheter bas, vendre haut”. Eh bien, quand les prix sont-ils absolument les plus bas ?
Quand tout le monde a peur de se pencher sur la situation et, comme le disait Rothschild, “quand le sang coule dans les rues”. Donc, c’est non seulement plus intéressant, mais c’est en fait moins risqué, pas plus risqué, car le risque est une question de prix. Et quand les prix sont bas, c’est moins risqué. Vous pouvez donc vous attendre à ce que je recherche des situations comme celle-ci à l’avenir.
Partout dans le monde, quelque part, à presque tout moment, il y a une bulle spéculative super ridicule en cours et, ailleurs, un marché à la baisse au plus bas de l’échelle qui atteint son paroxysme. Donc si vous regardez toutes ces choses, vous pouvez choisir ce qui convient à votre style d’investissement.
Nick Giambruno : Ok, Doug, parlons de certaines des fois où vous avez fait des investissements lorsque le sang coulait vraiment dans les rues.
Qu’en est-il de l’opportunité que vous avez eue d’acheter un château en Rhodésie (aujourd’hui Zimbabwe) ?
Doug Casey : C’était en 1978. Quoi qu’il en soit, j’ai écrit à ce sujet – et les chiffres sont exacts car je les ai réellement notés – dans la première édition de ma newsletter, qui s’appelait à l’époque Crisis Investing.
C’était à l’époque de la guerre. C’était vraiment la phase finale. Pourtant, lorsque vous arriviez dans le pays sur Air Rhodesia, vous deviez baisser les stores la nuit pour ne pas attirer les tirs anti-aériens.
Il y avait toutes sortes de choses qui se passaient. Et, vous savez, j’étais jeune et invulnérable. Je suis allé dans tout le pays, et j’étais le seul touriste, du moins le seul touriste qui n’était pas lourdement armé. J’étais la seule personne partout, des chutes Victoria aux ruines du Grand Zimbabwe. J’ai pris un bus à travers le pays, un petit mini-bus qui était en fait assez effrayant parce qu’ils tiraient sur les gens et tout ça.
Je voulais aller à Umtali, une ville à la frontière du Mozambique qui a été rebaptisée Mutare.
Quoi qu’il en soit, j’y suis allé, et l’endroit ressemblait à un camp militaire sorti de Mad Max, parce qu’il y avait tous ces véhicules blindés de fabrication artisanale qui circulaient.
Tout le monde m’a dit qu’il valait mieux aller voir l’hôtel Leopard Rock. C’est ce que j’ai fait, et c’était fantastique. C’était un château de 12 pièces que des prisonniers de guerre italiens avaient aidé à construire pendant la Seconde Guerre mondiale.
Il y avait 15 hectares de café et c’était magnifique, avec ces montagnes Bvumba qui surplombent le Mozambique… Il y avait un parcours de golf de neuf trous… Vous savez, tout ce que vous voulez dans un hôtel de villégiature.
J’aurais pu acheter cet endroit avec le linge, l’argenterie, tout pour 85 000 dollars.
Cela aurait fonctionné, car il s’est avéré que je suis retourné au Zimbabwe quelques années plus tard et qu’il venait de changer de mains pour 13,5 millions de dollars. Cela aurait donc été un joli coup.
Nick Giambruno : Parlez-nous de la fois où vous avez investi à Hong Kong pendant la crise chinoise de 1986.
Doug Casey : Cela a très bien fonctionné, en fait.
A cette époque, tout le monde pensait que les Chinois allaient prendre le contrôle de la ville. J’ai pu acheter un appartement penthouse dans un immeuble situé juste au-dessus du Hong Kong Yacht Club. Une vue fantastique sur le port, l’une des meilleures.
À l’époque, les gens m’ont dit que mon appartement en terrasse se vendait moins cher qu’un appartement au rez-de-chaussée, qui serait horrible à vivre avec tout le bruit de la rue. Mais la raison pour laquelle il se vendait si peu cher, c’est qu’ils étaient convaincus qu’il faudrait monter 13 étages à pied lorsque les Chinois prendraient le pouvoir, car ils ne répareraient pas les ascenseurs. Enfin, c’est comme ça que les choses fonctionnent.
J’ai acheté cet appartement pour quelque chose comme 40 000 dollars. C’est dire à quel point Hong Kong était bon marché à l’époque. Il m’a coûté 40 000 $ de plus, si je me souviens bien, pour le vider, le remeubler et le rendre très agréable.
Il y a quelques années, mon avocat m’a appelé et m’a parlé d’un appartement situé à l’étage inférieur de mon immeuble qui s’était vendu pour une somme ridicule.
Je lui ai dit : “Mettez-le sur le marché et faisons une offre demain matin.” Donc je pense que j’ai vendu cet endroit pour environ 1,2 millions de dollars. 80 000 $ à 1,2 million de dollars, un énorme retour sur investissement. En fait, j’ai été payé pour vivre là.
Et c’était un parfait exemple d’achat quand les gens avaient peur de Hong Kong. Ils avaient peur que les Chinois punissent les habitants de Hong Kong. Donc personne ne voulait être à Hong Kong, et en fait c’était le meilleur moment pour être à Hong Kong parce que c’était tellement bon marché.
Nick Giambruno : Merci pour ces histoires passionnantes, Doug.
Partout dans le monde, des personnes affirment que la pandémie est un mythe. C’est au Japon, en Suède, au Royaume-Uni et au Danemark que cette idée est la moins répandue, avec des résultats allant de trois à cinq pour cent. D’autre part, 30 % des personnes en Inde et 10% en France croient que “le coronavirus est un mythe produit par certaines forces puissantes et que le virus n’existe pas vraiment”.
Où les gens croient que le covid est un mythe
% disant que ce qui suit est vrai : “le coronavirus est un mythe créé par certaines forces puissantes et le virus n’existe pas vraiment”.
C’est la question utilisée lors d’un récent sondage YouGov visant à déterminer dans quelle mesure les gens du monde entier pensent ainsi.
En Inde, où 30 % des participants (urbains) partageaient cette opinion sur l’épidémie, la croyance en cette idée était de loin la plus populaire des 24 nations interrogées.