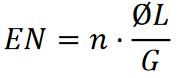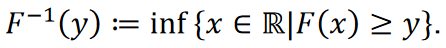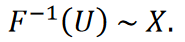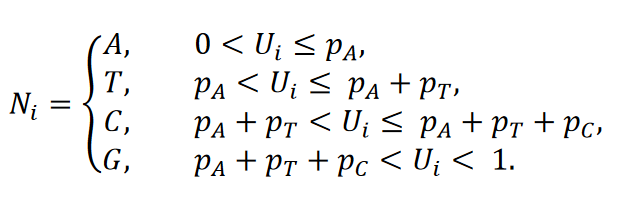TERRAIN expose la tyrannie de la fausse pandémie mondiale, fondée sur le modèle erroné de la maladie connu sous le nom de “théorie des germes”. Ce documentaire en deux parties explore la théorie du terrain, un modèle de santé qui fonctionne en symbiose avec la nature pour promouvoir le bien-être et la guérison, sans recourir à un paradigme médical corrompu et défectueux.
TERRAIN motive et inspire les spectateurs à comprendre le pouvoir et la responsabilité du consentement.
La première partie de TERRAIN remet en question la théorie des germes, un système de croyance obsolète et non scientifique basé sur des fraudes et des mauvaises interprétations.
La deuxième partie de TERRAIN explore les conséquences globales de l’adoption d’un modèle de santé non viable basé sur la théorie des germes et ouvre la porte à un biome synergique d’autocorrection et de guérison connu par tous les êtres vivants sous le nom de théorie du terrain.
Un film produit par Marcelina Cravat et Andrew Kaufman.
Avec le Dr Andrew Kaufman, le Dr Barre Lando, le Dr Stefan Lanka, le Dr Mark McDonald, le Dr Tom Cowan, le Dr Kelly Brogan, le Dr Samantha Bailey, Sayer Ji, Sally Fallon, Peggy Hall, Tony Roman, Alphonso Faggiolo et Veda Austin.
Un mathématicien allemand travaillant avec le Dr Stefan Lanka vient de publier un rapport intitulé “Analyse structurelle des données de séquençage en virologie – Une approche élémentaire à l’aide de l’exemple du SARS-CoV-2FR“. Il fournit encore plus de preuves que les virologues sont pris dans un monde de simulations informatiques – des simulations qui ne sont pas fiables même selon leurs propres termes, sans compter qu’elles sont déconnectées de la réalité. Cette analyse est une contribution importante qui expose un autre élément de l’anti-science utilisée pour soutenir cette fausse pandémie. En outre, il s’agit d’un démantèlement technique de la manière dont tous les “virus” sont inventés et ensuite “trouvés”, dans un jeu de tromperie permanent.
L’article est très technique et nécessite une certaine compréhension de la manière dont les virologues créent un “génome”, en partant d’un échantillon brut provenant d’un patient prétendument infecté par le virus “COVID-19”. Pour vous faciliter la tâche, j’ai produit un résumé des principales conclusions, présentées ci-dessous :
Il a été démontré qu’aucune des séquences génétiques utilisées pour produire les génomes du ” SARS-CoV-2 ” ne provenait de l’intérieur d’un virus. L’origine des fragments génétiques n’est pas claire.
La séquence originale de novo du ” SARS-CoV-2 ” construite par ordinateur et publiée par Fan Wu et al n’a pas pu être reproduite par la méthodologie décrite dans leur article, ce qui soulève des questions sur la façon dont ils l’ont produite et ont annoncé le nouveau ” virus ” au monde.
Les protocoles PCR sont calibrés sur des séquences d’origine non confirmée que l’on retrouve clairement chez de nombreux humains et apparemment aussi chez d’autres choses. Il n’a pas été démontré que le processus PCR permettait de détecter un “virus”, et encore moins de diagnostiquer une maladie inventée appelée “COVID-19”.
Les virologues se trompent eux-mêmes en effectuant des amplifications à 35 ou 45 cycles, car cela peut entraîner la “détection” de séquences qui ne sont même pas présentes dans l’échantillon. En effet, la méthodologie peut aboutir à la “détection” de n’importe quelle séquence qu’ils espèrent trouver.
Fan Wu et al auraient pu trouver de meilleures correspondances pour le “VIH” et le “virus de l’hépatite D” que pour “un nouveau coronavirus” chez leur homme de 41 ans de Wuhan, qui a présenté une pneumonie comme l’un des premiers cas déclarés de “COVID-19”. S’ils veulent trouver un “virus”, tout dépend de ce qu’ils demandent à l’ordinateur de chercher.
Bien sûr, cela a beaucoup plus de sens quand on s’attaque à la racine du problème : le ” SARS-CoV-2 ” n’est rien de plus qu’une simulation informatique et il n’y a jamais eu de virus à l’origine – tout cela est une fraude mondialeFR. La virologie semble ignorer qu’elle s’enfonce davantage dans une crise épistémologique, et pas seulement dans le domaine de la génomique, comme le souligne cet article de Mike Stone. Dans l’article de Stone, j’ai remarqué dans la section des commentaires que le Dr Valendar Turner du Perth Group a souligné que feu Sir John Maddox, ancien rédacteur en chef de Nature, avait lancé un avertissement pertinent en 1988. Il semble que ceux qui s’immergent dans le monde des techniques de détection moléculaire indirecte risquent de ne plus voir la forêt derrière les arbres, comme il le déclarait si justement :
“N’y a-t-il pas un danger, en biologie moléculaire, que l’accumulation de données prenne tellement d’avance sur leur assimilation dans un cadre conceptuel que les données finissent par constituer un obstacle ? Le problème vient en partie du fait que l’excitation de la chasse laisse peu de temps à la réflexion. Il y a des subventions pour produire des données, mais pratiquement aucune pour prendre du recul et réfléchir.”
Maddox, J. Nature 335, 11 (1998)
Nous nous efforcerons de continuer à dénoncerFR ces méthodologies antiscientifiques et d’encourager les autres à se demander si l’industrie de la virologie, qui représente des milliards de dollars, et les “traitements” bidon qui y sont associés et qui proviennent du gigantesque complexe pharmaceutique, aident réellement les gens à améliorer leur santé. Pour ceux d’entre nous qui voient qu’il n’y a aucune base solide à tout cela, il n’y a aucune chance que nous suivions les conseils des médecins et des scientifiques qui font la promotion de ces modèles malsains. Et, ce qui est peut-être encore plus important, nous savons qu’il ne faut prendre aucun des produits pharmaceutiques frauduleux et de plus en plus pervers qui sont le produit de cette pseudo-science et qui sont utilisés comme véhicules pour délivrer des composants néfastes et non répertoriés. Une fois de plus, vous pouvez éviter tous ces problèmes en indiquant :
Où est le virus* ?
*Particule minuscule qui est un parasite intracellulaire obligatoire (c’est-à-dire capable de se répliquer et transmissible) contenant un génome entouré d’une enveloppe protéique protectrice, codée par le virus.
Auteur : Dr. Mark Bailey
Mark est un chercheur dans le domaine de la microbiologie, de l’industrie médicale et de la santé qui a travaillé dans la pratique médicale, y compris les essais cliniques, pendant deux décennies.
La question de l’existence de virus pathogènes reste importante, car la croyance en de tels virus mobilise des milliards de dollars de ressources et de fonds de recherche. Ces deux dernières années, nous avons également vu comment un prétendu virus peut être utilisé comme un outil politique pour mettre les populations au pas. Ce n’est pas la première fois que cela se produit : par exemple, la “découverte” du VIH dans les années 1980 a donné naissance à une industrie de plusieurs milliards de dollars et a également été utilisée à des fins politiques dans la plupart des régions du monde. (Les erreurs concernant l’existence de la particule du VIH et le fait qu’elle soit à l’origine du sida sont décrites dans Virus ManiaFR. Pour ceux qui souhaitent approfondir le sujet, je recommande le magnus opus de The Perth Group sur ce sujet).
“Les virus sont de petits parasites intracellulaires obligatoires qui, par définition, contiennent un génome d’ARN ou d’ADN entouré d’une enveloppe protéique protectrice, codée par le virus.”
Medical Microbiology, 4th edition, 1996
Le journaliste indépendant Jeremy Hammond, qui se présente comme exposant la “dangereuse propagande d’Etat” entourant le COVID-19 et les dangers des vaccins, a ainsi fait la curieuse déclaration suivante en 2021 :
“l’affirmation fausse selon laquelle le SARS-CoV-2 n’a jamais été isolé (c’est-à-dire que son existence n’a jamais été prouvée) nuit considérablement à la crédibilité du mouvement pour la liberté de la santé et repose sur une ignorance totale de la science (le virus est constamment isolé et son génome entier est séquencé par des scientifiques du monde entier)”.
Jeremy Hammond, 9 mars 2021
Je dirais que l’ignorance est du côté de Hammond, qui semble parvenir à sa conclusion en répétant essentiellement les affirmations des virologues et en rassurant le public sur la validité de leurs méthodologies. Ces dernières semaines, nous avons également vu le Dr Joseph Mercola présenter l’interview de Hammond et le blog de Steve Kirsch (qui fait également appel à l’autorité de la virologie) comme des “preuves” de l’existence du SARS-CoV-2. Kirsch déclare s’appuyer sur “les avis des experts en qui j’ai confiance”, ce qui signifie qu’il a remis l’argument entre les mains d’autres personnes plutôt que d’enquêter lui-même sur la question. Mais est-il sage pour ces combattants de la liberté sanitaire qui s’opposent aux “experts” de l’establishment COVID de ne pas également remettre en question les virologues de l’establishment ?
Le Dr Andy Kaufman a produit une réfutation point par point du soutien de Hammond à la méthodologie d'”isolement” de la virologie moderne ici, tandis que le Dr Tom Cowan a prévenu que nous ne faisions que commencer à démanteler les absurdités de la virologie ici. Le Dr Sam Bailey a publié de nombreuses vidéos sur la question de l’isolement des virus, dont la plupart ont été interdites sur YouTube mais peuvent encore être trouvées sur Odysee. En outre, dans un essai que j’ai cosigné avec le Dr John Bevan-Smith, nous décrivons le premier pilier de la fraude COVID-19FR comme l’utilisation abusive du terme “isolement” par la virologie. En résumé, comme les virologues n’ont pas été en mesure d’isoler physiquement le moindre virus au siècle dernier, ils ont simplement changé la définition du mot, de sorte que même les virologues admettent que le terme est désormais utilisé de manière vague. Une situation étrange lorsque la méthode scientifique exige une terminologie précise.
J’ai observé au cours des deux dernières années que de nombreux scientifiques, médecins et journalistes sont heureux de sauter par-dessus ce gouffre de l'”isolement” et de citer les “génomes de coronavirus” déposés dans des bases de données comme preuve que le virus doit exister. Par exemple, Steve Kirsch écrit dans son blog que :
“Je sais que Sabine Hazan a vérifié que la séquence du virus obtenue auprès de l’ATCC correspondait exactement à ce qu’elle a trouvé chez les personnes atteintes du virus.”
Steve Kirsch, 10 janvier 2022
Il cite l’article de Hazan “Detection of SARS-CoV-2 from patient fecal samples by whole genome sequencing” comme preuve de cette affirmation. Kirsch admet qu’il ne sait pas comment les génomes ont été créés, mais ses…
“amis scientifiques semblent satisfaits avec eux. À 2 000 $ la dose, je ne pense pas qu’ils commercialiseraient le produit s’il était contaminé et inutile. Ai-je tort ?”
Steve Kirsch, 10 janvier 2022
Malheureusement, il semble avoir été dupé par la façade high-tech du génie génomique de la virologie, où des “virus” sont créés à partir de diverses séquences génétiques détectées. En fait, il arrive que les séquences ne soient pas vraiment détectées du tout, comme l’expose le Dr Stefan Lanka dans ce qui pourrait être le coup de grâce de la virologieFR.
L’article de Hazan peut servir d’exemple de la méthodologie défectueuse utilisée pour créer ces “génomes de virus”. L’équipe de recherche a obtenu des échantillons de matières fécales de 14 participants et a procédé à l’examen des séquences génétiques qu’elle pouvait détecter dans ces échantillons. Le premier problème se pose dans la section “méthodes”, lorsque l’équipe déclare que “le contrôle positif du SARS-CoV-2 de l’ATCC (SARS-CoV-2 inactivé par la chaleur, VR-1986HK ; souche 2019-nCoV/USA-WA1/2020) a été inclus tout au long du traitement de l’échantillon”. Comment ont-ils su que l’échantillon contenait le virus inactivé ? Parce que l’ATCC (American Type Culture Collection) l’affirme sur son site Web en déclarant que “cette souche a été isolée à l’origine d’un cas humain dans l’État de Washington et a été déposée par les Centers for Disease Control and Prevention”. Et comment les CDC ont-ils su qu’ils avaient le virus ? Parce qu’ils ont affirmé l’avoir trouvé dans cet article.
“Coronavirus 2 du syndrome respiratoire aigu sévère d’un patient atteint d’une maladie à coronavirus, États-Unis” Mais où était le virus ?
Dans le document des CDC, il est dit qu’ils ont recueilli “des spécimens cliniques d’un patient ayant contracté le COVID-19 lors d’un voyage en Chine et qui a été identifié à Washington, aux Etats-Unis”. Ils ont conclu que le patient avait le COVID-19 sur la base d’un résultat de PCR qui a détecté certaines séquences dites provenir du SARS-CoV-2. Mais à ce stade, ils n’avaient aucune preuve de l’existence d’un virus – tout ce qu’ils avaient, c’était quelques séquences génétiques détectées chez un patient atteint d’une infection virale présumée. Après avoir réalisé une expérience de culture tissulaire en tube à essai sur leur échantillon clinique et prétendu qu’il y avait des preuves de la présence d’un virus en raison d’effets cytopathiquesFR non spécifiques, ils ont commencé à construire leur “génome”. Ils déclarent que “nous avons utilisé 50 μL de lysat viral pour l’extraction de l’acide nucléique total pour les tests de confirmation et le séquençage.” Il s’agit d’un autre tour de passe-passe, car il n’a pas été démontré que le “lysat viral” provenait d’un virus, il s’agit simplement d’une soupe de cultures de cellules fragmentées et d’autres additifs.
L’affirmation selon laquelle ils ont “extrait l’acide nucléique des isolats” est tout aussi trompeuse. Ils ont laissé entendre qu’ils ont isolé un virus et qu’ils savent quelles séquences d’ARN proviennent de son contenu. Cependant, cela nécessiterait que les prétendues particules virales soient réellement isolées physiquement par purification, ce qu’ils n’ont pas fait. Et je dis “présumées” parce que même s’ils purifiaient les particules, il faudrait encore démontrer qu’elles répondent à la définition d’un virus – y compris le fait d’être un parasite et l’agent causal de la maladie – ce qui n’a pas été démontré par ces auteurs ni par aucun autreFR.
Dans tous les cas, comment ont-ils su quelles séquences génétiques appartenaient au “virus” en premier lieu ? Ils ont “conçu 37 paires de PCR emboîtées couvrant le génome sur la base de la séquence de référence du coronavirus (numéro d’accession GenBank NC045512)”. Et d’où vient cette “séquence de référence” ? Cela se rapporte à l’article de Fan Wu, et al décrivant l’homme de 41 ans qui a été admis à l’hôpital central de Wuhan le 26 décembre 2019 avec une pneumonie bilatérale et malgré l’absence de nouvelles caractéristiques cliniques, on a dit qu’il était atteint d’une maladie qui a ensuite été appelée “COVID-19”.
Le spécimen était constitué de lavages pulmonaires bruts, il contenait donc un mélange de cellules humaines et potentiellement toutes sortes d’autres micro-organismes et fragments génétiques. Ils ont simplement affirmé qu’il y avait un virus dans le mélange. À partir de cet échantillon mixte, ils ont généré à l’aveugle des dizaines de millions de séquences différentes, puis ont mis leur logiciel au travail pour voir comment ils pouvaient les assembler. Pour réaliser cet “ajustement”, le logiciel a recherché des “contigs”, c’est-à-dire des zones où différents fragments semblent avoir des séquences qui se chevauchent. Parmi les centaines de milliers de séquences hypothétiques générées de cette manière, ils ont constaté que la plus longue séquence “continue” que l’ordinateur a pu créer faisait environ 30 000 bases et ont conclu que cette création informatique devait être le génome du nouveau virus présumé.
Ils pensaient qu’il s’agissait du génome parce que leur séquence de 30 000 bases générée de manière hypothétique était similaire à 89,1 % à ” un isolat de coronavirus (CoV) de chauve-souris semblable au SRAS, le SL-CoVZC45 “. Le “génome” de l'”isolat” de CoV de chauve-souris a été généré en 2018 après que “19 paires d’amorces PCR dégénérées ont été conçues par alignement multiple des séquences SARS-CoV et SL-CoV de chauve-souris disponibles déposées dans GenBank, ciblant presque toute la longueur du génome.” En d’autres termes, ils connaissaient déjà la séquence à rechercher sur la base des séquences qui avaient été précédemment déposées dans la GenBank. Mais comment les producteurs de ces séquences déjà déposées savaient-ils qu’ils avaient trouvé des génomes viraux ? Bienvenue dans le raisonnement circulaire de la virologie moderne.
Pour expliquer la boucle dans laquelle les virologues semblent être pris au piège, cet article de 2019 publié dans Virology illustre bien le problème :
“Trois méthodes principales basées sur le HTS [séquençage à haut débit] sont actuellement utilisées pour le séquençage du génome entier viral : le séquençage métagénomique, le séquençage par enrichissement de cible et le séquençage par amplicon PCR, chacune présentant des avantages et des inconvénients (Houldcroft et al., 2017). Dans le séquençage métagénomique, l’ADN (et/ou l’ARN) total d’un échantillon comprenant l’hôte mais aussi des bactéries, des virus et des champignons est extrait et séquencé. C’est une approche simple et rentable, et c’est la seule approche qui ne nécessite pas de séquences de référence. Au contraire, les deux autres approches HTS, l’enrichissement des cibles et le séquençage des amplicons, dépendent toutes deux d’informations de référence pour concevoir les appâts ou les amorces.”
Maurier F, et al, “A complete protocol for whole-genome sequencing of virus from clinical samples,” Virology, May 2019.
On touche là à la racine du problème. Les génomes de référence “viraux” sont créés par séquençage métagénomique, mais celui-ci est effectué sur des spécimens bruts (tels que des lavages de poumons ou des cultures de tissus non purifiés) et l’on déclare ensuite que les séquences sélectionnées sont d’origine virale. Il y a donc déjà deux problèmes : premièrement, il n’y a pas eu d’étape (c’est-à-dire de purification) pour montrer que les séquences proviennent de l’intérieur de “virus” et deuxièmement, comme décrit ci-dessus, les “génomes” générés par ordinateur sont simplement des modèles hypothétiques assemblés à partir de petits fragments génétiques, et non quelque chose dont l’existence a été prouvée dans la nature comme une séquence entière de 30 000 bases. Cependant, ces modèles in silico deviennent alors effectivement le “virus” et une entité telle que le SARS-CoV-2 est créée. Une fois que la première séquence de ce type est déposée dans une base de données, le “virus” peut être “trouvé” par d’autres grâce aux mêmes techniques métagénomiques défectueuses. Ou, comme l’indique l’article de Virology, il peut être “trouvé” par enrichissement de la cible et séquençage de l’amplicon (généralement par PCR), mais cela nécessite de disposer d’une séquence de référence… c’est-à-dire d’un modèle inventé in silico par séquençage métagénomique où la provenance des fragments génétiques était inconnue.
Il n’y a aucune partie dans le processus ci-dessus qui établit soit :
1) la composition génétique de toute particule imagée ou imaginée ; ou
2) la nature biologique de ces particules, c’est-à-dire ce qu’elles font réellement.
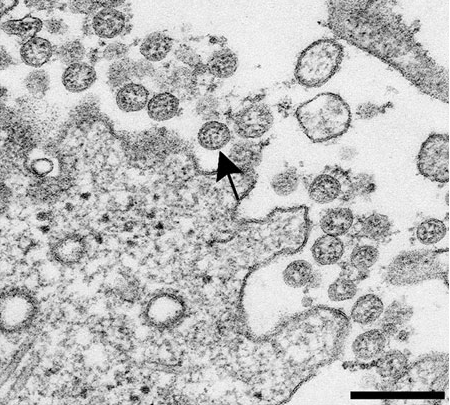
C’est une belle nanoparticule, mais de quoi est-elle faite et que fait-elle ?
Pouvons-nous maintenant revenir à l’article de Hazan pour constater qu’il s’agit d’un exercice inutile de virologie absurde. Ils déclarent qu’avec leur “contrôle positif du SARS-CoV-2 provenant de l’ATCC”, les “génomes des patients ont été comparés au génome de référence du SARS-CoV-2 Wuhan-Hu-1 (MN90847.3)”. Le numéro d’accès MN90847.3 fait référence au “génome” actualisé qui aurait été trouvé chez l’homme de 41 ans de Wuhan, comme indiqué ci-dessus dans l’article de Fan Wu et al. La boucle est bouclée : à aucun moment il n’a été démontré qu’il y avait un virus en suivant cette piste de “génomes”. L’équipe de Fan Wu n’a jamais trouvé de virus, elle a simplement affirmé que sa simulation informatique de séquence génétique était une “nouvelle souche de virus à ARN de la famille des Coronaviridae”, sans prouver que la séquence existait dans la nature ou provenait de l’intérieur d’un virus. Par conséquent, il n’y a pas eu de “détection du SARS-CoV-2 à partir d’échantillons de matières fécales de patients” comme le prétend le titre de l’article de Hazan, à moins que “SARS-CoV-2” ne signifie des séquences génétiques d’on-ne-sait-quoi provenant d’on-ne-sait-où. Peu importe où ou à quelle fréquence ces séquences sont détectées – il n’a jamais été prouvé qu’elles étaient de nature virale. Ainsi, lorsque Steve Kirsch affirme que Hazan “a vérifié que la séquence du virus obtenue de l’ATCC correspondait exactement à ce qu’elle a trouvé chez les personnes atteintes du virus”, il se trompe.
De quel “virus” parle-t-il ?
Auteur : Dr. Mark Bailey
Mark est un chercheur dans le domaine de la microbiologie, de l’industrie médicale et de la santé qui a travaillé dans la pratique médicale, y compris les essais cliniques, pendant deux décennies.
Source (en anglais) : https://drsambailey.com/covid-19/warning-signs-youve-been-tricked-by-virologists/
Stefan Lanka, en collaboration avec un mathématicien anonyme, vient de rendre publiques ses recherches sur l’analyse du génome du SARS-CoV-2 et des techniques et méthodes questionnables utilisées par les virologues.
C’est l’un des derniers éléments qu’il manquait pour complètement réfuter, méthodiquement, toutes allégations d’un nouveau virus contagieux responsable d’une nouvelle maladie tel que décrit et accepté actuellement.
Pour résumer simplement cette étude assez technique, la publication à l’origine du premier génome du SARS-CoV-2 n’est pas reproductible, car les données fournies ne permettent pas d’aboutir aux mêmes résultats.
Il est également démontré que les données publiées ont été manipulées (dans le sens “travaillées”) et qu’elles ne correspondent pas à ce qu’elles devraient normalement représenter tel que revendiqué.
Ce simple article, sur la base de différentes études virologiques et à l’aide de plusieurs outils bio-informatiques, permet de remettre en cause l’ensemble du bien-fondé du domaine de la génétique appliquée à la virologie moderne, et donc du SARS-CoV-2, le virus présumé responsable de la maladie Covid-19, dont la réalité repose presque exclusivement sur ces méthodes et techniques.
Analyse structurelle des données de séquençage en virologie
Une approche élémentaire à partir de l’exemple du SARS-CoV-2
Auteur
Par un mathématicien de Hambourg, qui souhaite rester anonyme.
Abstract
Le séquençage méta-transcriptomique de novo ou le séquençage du génome entier sont des méthodes acceptées en virologie pour la détection de prétendus virus pathogènes. Dans ce processus, aucune particule virale (virion) n’est détectée et, au sens du mot isolement, isolée et caractérisée biochimiquement. Dans le cas du SARS-CoV-2, l’ARN total est souvent extrait d’échantillons de patients (par exemple : liquide de lavage broncho-alvéolaire (LBA) ou écouvillons de gorge) et séquencé. Notamment, il n’y a aucune preuve que les fragments d’ARN utilisés pour le calcul des séquences du génome viral soient d’origine virale.
Nous avons donc examiné la publication “A new coronavirus associated with human respiratory disease in China” [1] et les données de séquence publiées associées avec le bioprojet ID PRJNA603194 daté du 27/01/2020 pour la proposition de séquence génétique originale du SARS-CoV-2 (GenBank : MN908947.3). Une répétition de l’assemblage de novo avec Megahit (v.1.2.9) a montré que les résultats publiés ne pouvaient pas être reproduits. Nous avons peut-être détecté des acides ribonucléiques (ribosomaux) d’origine humaine, contrairement à ce qui a été rapporté dans [1]. Une analyse plus poussée a fourni des preuves d’une possible amplification non spécifique des lectures pendant la confirmation par PCR et la détermination des terminaisons génomiques non associées au SARS-CoV-2 (MN908947.3).
Enfin, nous avons réalisé des assemblages de référence avec des séquences génomiques supplémentaires telles que le SARS-CoV, le virus de l’immunodéficience humaine, le virus de l’hépatite delta, le virus de la rougeole, le virus Zika, le virus Ebola ou le virus de Marburg, afin d’étudier la similarité structurelle des données de séquence actuelles avec les séquences respectives. Nous avons obtenu des indications préliminaires selon lesquelles certaines des séquences du génome viral que nous avons étudiées dans le présent travail pourraient être obtenues à partir de l’ARN d’échantillons humains insoupçonnés.
Mots-clés
SARS-CoV-2, COVID-19, Virus, assemblage de novo, séquençage du génome entier, WGS, bioinformatique, PCR, SARS-CoV, Bat SARS-CoV, virus de l’immunodéficience humaine, VIH, virus de l’hépatite, virus de la rougeole, virus Zika, virus Ebola, virus de Marburg.
Introduction
Pour construire des séquences génomiques virales, les acides nucléiques (ARN ou ADN) sont isolés à partir de diverses sources d’acides nucléiques telles que le liquide de lavage broncho-alvéolaire (LBA) [1, 2], les écouvillons nasopharyngés [3, 4, 5, 6, 12, 13], les composants de culture cellulaire ou les surnageants de culture cellulaire [2, 11, 12, 13, 14, 16], ainsi qu’à partir d’échantillons humains [8, 9, 10, 16] et animaux [7, 15], puis séquencés. Dans ce processus, les acides nucléiques obtenus ne proviennent pas exclusivement de particules (de virus) préalablement isolées, c’est-à-dire séparées de tout le reste, mais souvent de l’échantillon entier. Ainsi, l’origine des fragments d’acide nucléique utilisés pour calculer les séquences génomiques n’est a priori pas claire.
Dans le cas des acides ribonucléiques (ARN), ceux-ci sont d’abord transcrits en ADNc à l’aide d’une ADN polymérase ARN-dépendante. L’ADN ou l’ADNc est ensuite fragmenté à l’aide d’enzymes et amplifié par réaction en chaîne par polymérase (PCR) avant que le séquençage proprement dit, c’est-à-dire la détermination de la séquence nucléotidique des courts fragments d’ADN ou d’ADNc, n’ait lieu. Lors de l’amplification, outre des séquences d’amorces aléatoires (hexamères aléatoires), des séquences d’amorces hautement spécifiques sont également utilisées en fonction des génomes de référence ou des génomes cibles considérés [par exemple : 1, 3, 4, 5, 6, 7, 8, 17, 18]. Enfin, les données de séquence ainsi obtenues sont traitées à l’aide d’algorithmes bioinformatiques.
Deux méthodes courantes pour déterminer les séquences du génome viral sont l’assemblage méta-transcriptomique de novo [1, 12] et le séquençage du génome entier (whole genome sequencing) [3, 4, 5, 6, 17, 18]. Alors que l’assemblage méta-transcriptomique de novo n’utilise souvent aucune séquence de référence ou seulement des séquences de référence en aval, le séquençage du génome entier utilise un grand nombre de séquences d’amorces spécifiques, dont certaines couvrent déjà ensemble 4 à 17 % du génome cible [1, 17]. Pour l’amplification de l’ADNc, 35 à 45 cycles sont souvent utilisés [1, 6, 17].
Dans le cas du SARS-CoV-2 (GenBank : MN908947.3) [1], la proposition de séquence du génome viral a été calculée par assemblage méta-transcriptomique de novo de l’ARN total provenant du LBA (lavage broncho-alvéolaire) d’un patient de Wuhan, en Chine. Les assembleurs Megahit (v.1.1.3) et Trinity (v.2.5.1) ont été utilisés pour assembler les contigs. Megahit a généré un total de 384.096 (200 nt – 30.474 nt) et Trinity a calculé 1.329.960 (201 nt – 11.760 nt) contigs. Les grandes différences entre les deux assemblages sont notables. Selon [1], le plus long contig assemblé avec Megahit présentait une grande similarité nucléotidique (89,1%) avec le génome de la chauve-souris SL-CoVZC45 (GenBank : MG772933) et a été utilisé pour concevoir des amorces pour la confirmation par PCR et les terminaisons du génome.
L’organisation du génome viral a été déterminée par alignement de séquences sur deux espèces représentatives du genre Betacoronavirus, un coronavirus associé à l’homme (SARS-CoV Tor 2, GenBank : AY274119) et un coronavirus associé aux chauves-souris (bat SL-CoVZC45, GenBank : MG772933).
Aucune particule virale pathogène associée de manière unique à la séquence MN908947.3 n’a été identifiée et caractérisée biochimiquement à partir de l’échantillon du patient. Au contraire, l’ARN total a été extrait et traité à partir du LBA d’un patient. Il n’y a pas de preuve que seuls des acides nucléiques viraux ont été utilisés pour construire le génome viral revendiqué pour le SARS-CoV-2. De plus, en ce qui concerne la construction du brin de génome viral revendiqué, aucun résultat d’éventuelles expériences témoins n’a été publié. Ceci est également vrai pour toutes les autres séquences de référence considérées dans le présent travail. Dans le cas du SARS-CoV-2, un contrôle évident serait que le génome viral revendiqué ne puisse pas être assemblé à partir de sources d’ARN non suspectées d’origine humaine, ou même d’autres origines.
Dans la présente publication, nous avons étudié la reproductibilité des assemblages de novo en utilisant les données de séquence originales publiées pour le travail original sur le coronavirus SARS-CoV-2 [1]. Nous avons également étudié la similarité structurelle des données de séquence actuelles avec d’autres séquences virales de référence accessibles au public pour le SARS-CoV (chauve-souris) [1, 7, 13, 14], le virus de l’immunodéficience humaine [8], le virus de l’hépatite delta [9], le virus de la rougeole [11, 12], le virus Zika [10], le virus Ebola [15] et le virus de Marburg [16] (Tableaux et Figures : Tableau 3). A cette fin, nous présentons ici un protocole bioinformatique simple. Pour valider nos résultats, nous avons également considéré des séquences génomiques générées de manière aléatoire et fictive afin d’exclure le caractère purement aléatoire de nos résultats.
Section principale
Reconstitution de l’assemblage de novo des données de séquence publiées
Pour répéter l’assemblage de novo, nous avons téléchargé les données de séquence originales (SRR10971381) du 27/01/2020 au 11/30/2021 en utilisant les outils SRA [19] à partir d’Internet. Pour préparer les lectures en paires pour l’étape d’assemblage avec Megahit (v.1.2.9) [20], nous avons utilisé le préprocesseur FASTQ fastp (v.0.23.1) [21]. Après avoir filtré les lectures en paires, 26 108 482 des 56 565 928 lectures initiales sont restées, avec une longueur d’environ 150 pb. Une grande partie des séquences, vraisemblablement une majorité de celles d’origine humaine, ont été écrasées par les auteurs avec “N” pour inconnu et donc filtrées par fastp. Ceci doit être considéré comme un problème au sens de la scientificité, puisque toutes les étapes ne peuvent pas être retracées ou reproduites. Pour la génération élaborée de contigs à partir des lectures de séquences courtes restantes, nous avons utilisé Megahit (v.1.2.9) en utilisant les paramètres par défaut.
Nous avons obtenu 28 459 (200 nt – 29 802 nt) contigs, soit beaucoup moins que ce qui est décrit dans [1]. Contrairement aux représentations de [1], le contig le plus long que nous avons assemblé ne comprenait que 29 802 nt, soit 672 nt de moins que le contig le plus long avec 30 474 nt, qui selon [1] comprenait presque tout le génome viral. Notre contig le plus long a montré une correspondance parfaite avec la séquence MN908947.3 à une longueur de 29 801 nt (Tableaux et Figures, Tableaux 1, 2). Nous n’avons donc pas pu reproduire le contig le plus long de 30 474 nt, qui est si important pour la vérification scientifique. Par conséquent, les données de séquence publiées ne peuvent pas être les lectures originales utilisées pour l’assemblage.
Après avoir assemblé les contigs, nous avons déterminé la richesse de couverture respective en faisant correspondre les séquences courtes aux 28 459 contigs déterminés à l’aide de Bowtie2 (v.2.4.4) [22]. Nous avons ensuite fait correspondre les 50 contigs ayant la plus grande abondance de couverture et les 50 contigs les plus longs à la base de données de nucléotides (Blastn) le 12/05/2021 et le 12/20/2021, respectivement. Les résultats détaillés des requêtes se trouvent dans les tableaux et figures : Tableaux 1, 2.
Une comparaison de nos résultats (Tables and Figures: Table 1) avec ceux de [1, Supplementary Table 1. The top 50 abundant assembled contigs generated using the Megahit program.] montre des différences remarquables. Dans ce qui suit, les ID de contigs de [1] sont précédés de “1_” pour mieux les distinguer de nos ID de contigs. En général, on peut dire que les résultats de nos requêtes concernant les numéros d’accession ne correspondent pas exactement à ceux de [1]. En ce qui concerne les descriptions des sujets, nous avons observé une bonne correspondance pour la plupart. De plus, à l’exception du contig le plus long (1_k141_275316), nos contigs se sont avérés plus longs et ont eu tendance à avoir une couverture plus riche. Le cas est clair pour le contig 1_k141_179411 comparé au contig k141_12253. Le premier a une longueur de 2 733 nt, tandis que le second a une longueur de 5 414 nt. Cela fournit la première indication possible que l’amplification non spécifique de lectures de séquences non associées au SARS-CoV-2 s’est produite pendant la confirmation par PCR avec des amorces construites pour MN908947.3 à partir de 1_k141_275316 (30,474 nt).
À ce stade, le contig avec l’identification k141_27232, auquel 1 407 705 séquences sont associées, et donc environ 5% des 26 108 482 séquences restantes, doit être discuté en détail. L’alignement avec la base de données de nucléotides le 05/12/2021 a montré une correspondance élevée (98,85%) avec “Homo sapiens RNA, 45S pre-ribosomal N4 (RNA45SN4), ribosomal RNA” (GenBank : NR_146117.1, daté du 04/07/2020). Cette observation contredit l’affirmation de [1] selon laquelle la déplétion de l’ARN ribosomal a été effectuée et les lectures de séquences humaines ont été filtrées à l’aide du génome de référence humain (version 32 humaine, GRCh38.p13). Il convient de noter que la séquence NR_146117.1 n’a été publiée qu’après la publication de la bibliothèque de séquences SRR10971381 considérée ici.
Cette observation souligne la difficulté de déterminer a priori l’origine exacte des fragments individuels d’acide nucléique utilisés pour construire les séquences génomiques virales revendiquées.
Analyse de la structure des séquences basée sur les références
Nous avons d’abord mappé les lectures en paires (2×151 pb) avec BBMap [23] aux séquences de référence que nous avons considérées (Tableaux et Figures : Tableau 3) en utilisant des paramètres relativement peu spécifiques. Nous avons ensuite fait varier la longueur minimale (M1) et l’identité (nucléotidique) minimale (M2) avec reformat.sh pour obtenir des sous-ensembles correspondants des séquences précédemment cartographiées avec une qualité appropriée. L’augmentation de la longueur minimale M1 ou de l’identité nucléotidique minimale M2 augmente ainsi la significativité de la cartographie respective. Ensuite, nous avons formé des séquences consensus avec les sous-ensembles respectifs de qualité sélectionnée par rapport à la référence sélectionnée. Nous avons attribué la valeur “N” (inconnu) à toutes les bases dont la qualité est inférieure à 20. Une qualité de 20 signifie un taux d’erreur de 1% par nucléotide, ce qui peut être considéré comme suffisant dans le contexte de nos analyses. Enfin, l’évaluation de la concordance entre les séquences de référence et les séquences consensus a été réalisée à l’aide de BWA [24], Samtools [25] et Tablet [26]. La paire ordonnée (M1 ; M2) = (37 ; 0.6) a été juste choisie pour donner des taux d’erreur F1 et F2, respectivement, de moins de 10% pour la référence LC312715.1. Les résultats de tous les calculs effectués sont présentés dans les tableaux et les figures : Tableau 4. Les calculs montrent la signification la plus élevée pour le choix de la paire ordonnée (37 ; 0.6), ce qui peut être vu par les taux d’erreur les plus élevés dans chaque cas. Une signification comparable est fournie par les paires ordonnées (47 ; 0,50) et (25 ; 0,62). Alors que les séquences génomiques associées aux coronavirus présentent des taux d’erreur approximativement supérieurs à 10% pour toutes les paires ordonnées considérées (M1 ; M2), les taux d’erreur des deux séquences LC312715.1 (VIH) et NC_001653.2 (Hépatite delta) sont inférieurs à 10% et diminuent encore pour les paires ordonnées (32 ; 0,60) et (30 ; 0,60). La séquence MG772933_short est principalement constituée de la partie qui n’est pas recouvrable par les lectures associées au SARS-CoV-2 (voir Tableaux et Figures : Figure 3). Là encore, aucune amélioration n’a pu être obtenue en réduisant les valeurs de M1 et M2. Les taux d’erreur des séquences NC_039345.1 (virus Ebola), NC_024781.1 (virus Marburg), AF266291.1 et KJ410048.1 (virus de la rougeole) sont nettement plus élevés que ceux des séquences LC312715.1 et NC_001653.2. Alors que les séquences d’acides nucléiques utilisées pour calculer les premiers génomes ont été propagées dans des cellules Vero, les séquences d’acides nucléiques utilisées pour LC312715.1 et NC_001653.2 proviennent directement d’échantillons d’origine humaine (Tableaux et Figures : Tableau 3). Par conséquent, la question se pose de savoir si ce résultat est dû à des différences structurelles des sources d’acides nucléiques respectives ou aux protocoles de séquençage respectifs utilisés. Par exemple, la transcriptase inverse utilisée pour convertir l’ARN en ADNc ou les séquences d’amorces utilisées pour l’amplification ainsi que les cycles d’amplification pourraient éventuellement entraîner des différences dans les bibliothèques de séquences obtenues.
Les taux d’erreur F1 et F2 les plus élevés sont affichés par les séquences génomiques fictives générées aléatoirement rnd_uniform, rnd_wuhan, rnd_wh_mk_1 et rnd_wh_mk_2, de sorte que les résultats trouvés ici ne sont pas purement aléatoires.
Analyse graphique des distributions de couverture et des longueurs de lecture
Après avoir observé la possibilité de former des séquences consensus de haute qualité par rapport à certaines séquences de référence, nous avons analysé la distribution de la couverture des lectures de courtes séquences associées (Tableaux et Figures : Figures 1-22) et la distribution des longueurs de lecture (Tableaux et Figures : Figures 23-25). Pour ce faire, nous avons préalablement mappé les lectures de séquences courtes à leurs séquences de référence respectives en utilisant BBMap, [(M1 ; M2) = (37 ; 0.60)]. En plus des séquences courtes, nous avons également mappé les 26 paires d’amorces [1, Supplementary Table 8. PCR primers used in this study.] pour le séquençage du génome entier du SARS-CoV-2 (GenBank : MN908947.3) aux génomes de référence considérés. L’analyse subséquente a été effectuée via Tablet et le tableur Excel.
Tout d’abord, nous considérons la référence générée aléatoirement rnd_uniform. Des observations comparables s’appliquent aux génomes de référence générés aléatoirement rnd_wuhan, rnd_wh_mk_1 et rnd_wh_mk_2 (Tableaux et Figures : Figures 14-16).
Figure 13 : Référence rnd_uniform. a) rnd_uniform_reads cartographié à l’aide de BBMap, (M1 ; M2) = (37 ; 0,60). b) rnd_uniform_primer cartographié à l’aide de BBMap. c) La couverture distribuée exponentielle a été générée par simulation stochastique à l’aide de la méthode d’inversion. d) Les 26 paires d’amorces ([1, Tableau supplémentaire 8. Amorces PCR utilisées dans cette étude]) sont réparties de manière inégale sur l’ensemble du génome de référence. Les positions des amorces ne sont que faiblement corrélées avec les zones de couverture nucléotidique élevée, chacune ne comprenant que quelques nucléotides. e) La distribution des rnd_uniform_reads semble largement aléatoire. La variance de la distribution exponentielle considérée correspond bien à la variance empirique ajustée.
La couverture (rnd_uniform_reads) varie de manière aléatoire et relativement homogène sur toutes les positions nucléotidiques. La structure est comparable à celle de la couverture générée aléatoirement (couverture distribuée exponentielle), bien que la variance semble un peu plus faible. À quelques positions nucléotidiques isolées, la couverture présente une couverture élevée par rapport à la moyenne, mais chacune d’entre elles ne couvre que quelques régions nucléotidiques contiguës. Une corrélation avec les positions des amorces n’est que faiblement prononcée. La couverture d’apparence purement aléatoire avec les lectures de séquences courtes est corrélée avec une séquence consensus mappable non continue et un taux d’erreur F1 élevé de 38,60 %. Ainsi, la structure aléatoire (interne) des nucléotides de la séquence de référence simulée stochastiquement “rnd_uniform” est relativement absente des données de séquence examinées ici.
Par contraste, nous considérons maintenant le génome de référence du SARS-CoV-2 (GenBank : MN908947.3).
Figure 1 : Référence MN908947.3. a) MN908947_reads mappé avec Bowtie2 en utilisant les paramètres par défaut. b) MN908947_primer cartographié à l’aide de BBMap. c) Les quantiles ont été déterminés à partir de EN et VARN sous l’hypothèse de distribution d’une distribution binomiale. d) Les 26 paires d’amorces ([1], Tableau supplémentaire 8. Amorces PCR utilisées dans cette étude.) sont réparties uniformément sur l’ensemble du génome de référence. Les positions des amorces sont en corrélation avec les zones de couverture nucléotidique élevée.
Contrairement à la figure 13, la distribution de la couverture présente davantage un modèle en forme de vague avec des couvertures de nucléotides régulières et significativement plus importantes. Les 26 paires d’amorces sont réparties uniformément sur toutes les positions nucléotidiques de la séquence de référence. Les positions d’amorce sont souvent situées près des positions nucléotidiques avec une couverture nucléotidique élevée par rapport à la moyenne. Cela indique que toutes les parties du génome de référence n’ont pas été amplifiées de manière égale. En supposant que les 29 903 positions nucléotidiques ont la même probabilité d’apparaître dans les lectures associées au SARS-CoV-2, la couverture de chaque position nucléotidique devrait se situer entre les deux lignes avec une probabilité de 99,5 % (en supposant une distribution binomiale). Ce n’est pas le cas pour environ 90% des positions nucléotidiques. A priori, on pourrait s’attendre à ce que si une quantité suffisante d’ARN viral est présente dans l’échantillon et que suffisamment de morceaux de séquence sont lus, on obtienne une couverture homogène des nucléotides au sein du génome viral.
Le graphique suivant permet d’étudier les distributions des longueurs de lecture des références que nous venons de considérer (rnd_uniform et MN908947.3)
Figure 23 : a)-f) Cartographie à l’aide de BBMap, (M1 ; M2) = (37 ; 0,60). Analyse dans Excel.
La figure 23e) montre la distribution des longueurs de lecture dans le cas de la référence “rnd_uniform”. La longueur moyenne des lectures est de 41,96 nt, à peine à droite du maximum de la distribution. En comparaison, la distribution pour la référence MN908947.3, Figure 23a) montre une région proéminente (aléatoire) similaire à la Figure 23e) et une région distincte avec des lectures d’environ 150 nt de longueur. La longueur moyenne des lectures est supérieure à 110 nt. Toutes les séquences de référence avec une distribution comparable et donc plutôt aléatoire des longueurs de lecture comme dans la référence “rnd_uniform” simulée de manière stochastique (Tableaux et Figures : Figure 23d), f) ; Figure 24d), e), f) ; Figure 25a) – c)) présentent également des taux d’erreur élevés F1 et F2 (Tableaux et Figures : Tableau 4).
Cette constatation est mise en évidence par l’analyse suivante. Afin de mieux comprendre la structure interne des quelque 56 millions de séquences publiées, nous avons considéré la condition supplémentaire maxlength=100 pour la séquence MN908947.3 lors de la formation des sous-ensembles après cartographie avec BBMap en plus de M1 et M2.
Figure 2 : Référence MN908947.3. a) MN908947_reads cartographié avec Bowtie2 en utilisant les paramètres par défaut. b) MN908947_short_reads cartographié avec BBMap, (M1 ; M2) = (37 (max. 100) ; 0.60). c) La couverture distribuée exponentielle a été générée par simulation stochastique en utilisant la méthode d’inversion. La distribution de la couverture MN908947_short_reads présente un modèle plus aléatoire, mais sa variance ajustée est plus élevée. Ceci est principalement dû aux quelques fluctuations de la distribution de la couverture.
En excluant toutes les séquences mappables de plus de 100 nucléotides, on a essentiellement éliminé les quelque 120 000 lectures associées au SARS-CoV-2. La distribution de la couverture des courtes séquences restantes semble maintenant aléatoire, de façon analogue à la figure 13. Là encore, cela correspond aux taux d’erreur élevés de R1 (29,90 %) et R2 (29,96 %). Cela indique qu’aucune structure significative de la référence MN908947.3 n’est incluse dans les séquences publiées, à l’exception des quelque 120 000 (tableaux et figures. Tableau 1) lectures courtes associées.
Avant d’entrer dans le détail de certains des génomes de référence que nous avons examinés, nous aimerions d’abord examiner la couverture de deux autres contigs : k141_12253 et k141_20796. Alors que le contig identifié comme k141_12253 est caractérisé par une couverture relativement élevée, k141_20796 fait partie des trois plus longs contigs calculés.
Figure 18 : Référence k141_12253. a) k141_12253_reads mappé avec Bowtie2 en utilisant les paramètres par défaut. b) k141_12253_primer cartographié avec BBMap.
Le contig k141_12253 présente une grande similarité avec la bactérie Leptotrichia (GenBank : CP012410.1). Sur les 52 séquences d’amorces publiées, 38 ont pu être cartographiées sur la référence k141_12253 avec un taux d’erreur relativement élevé de 37,30%. La distribution de la couverture s’avère être extrêmement inhomogène et montre, surtout dans les 500 premiers nucléotides, une couverture nucléotidique extrêmement élevée par rapport à la moyenne. Les zones présentant une couverture élevée sont en corrélation avec les positions d’amorce déterminées. Cela pourrait indiquer que des lectures non exclusivement associées au SARS-CoV-2 ont été amplifiées en grande quantité. Compte tenu du taux d’erreur relativement élevé de 37,30 %, cela impliquerait une amplification relativement non spécifique. Ainsi, la question se pose de savoir si les lectures obtenues par l’amplification de l’ADNc avec les séquences d’amorce spécifiques étaient déjà présentes dans l’échantillon initial ou ont été générées par la procédure elle-même.
Figure 21 : Référence k141_20796. a) k141_20796_reads mappé avec Bowtie2 en utilisant les paramètres par défaut. b) k141_20796_primer cartographié avec BBMap.
Le contig k141_20796, qui a une correspondance élevée avec la bactérie Veillonella parvula (GenBank : LR778174.1), montre une couverture plus faible avec des lectures associées par rapport au contig avec l’identification k141_12253. La structure de la couverture nucléotidique est similaire à celle du SARS-CoV-2 (GenBank : MN908947.3). Notamment, la couverture est à nouveau inhomogène, ce qui indique une amplification inégale. En raison de la longueur nucléotidique plus élevée, 47 des 52 séquences d’amorces publiées ont pu être cartographiées sur le contig de référence avec un taux d’erreur moyen de 35,80 %. Encore une fois, les positions des amorces sont bien corrélées avec les zones de couverture nucléotidique élevée. Cela pourrait à nouveau indiquer une amplification non spécifique de séquences non associées au SARS-CoV-2 (GenBank : MN908947.3).
Dans la présente section, nous examinerons plus en détail les séquences de référence “Virus de l’immunodéficience humaine 1” (GenBank : LC312715.1) et “Virus de la rougeole de génotype D8 souche MVi/Muenchen” (GenBank : KJ410048.1). Toutes les autres figures se trouvent dans les documents supplémentaires (Tableaux et Figures : Figures 1-22 et Figures 23-25).
Figure 6 : Référence LC312715.1. a) LC312715.1_short_reads cartographié en utilisant BBMap, (M1 ; M2) = (37 ; 0.60). b) LC312715.1_primer cartographié en utilisant BBMap.
Déjà dans la section précédente, une grande similarité structurelle des séquences publiées avec la séquence de référence LC312715.1 a été montrée. La séquence consensus calculée a montré des taux d’erreur relativement plus faibles R1 = 8,60 % et R2 = 8,83 % en comparaison avec, par exemple, les références associées au SRAS. La figure 6 montre de nettes différences avec la figure 13. La distribution de la couverture montre également plus un modèle en forme de vague avec des zones relativement régulières de couverture particulièrement élevée et est donc clairement différente de la distribution de la couverture de la référence aléatoire “rnd_uniform”. La distribution des longueurs de lecture (Figure 23b), comparer également c)) diffère également de manière significative des distributions plus aléatoires et montre un nombre significatif de lectures cartographiables avec des longueurs allant jusqu’à environ 110 nt. La longueur moyenne des lectures de 51,84 nt est également plus élevée que pour “rnd_uniform”, par exemple.
Une fois encore, il est intéressant de noter la position des séquences d’amorce par rapport aux zones de couverture nucléotidique élevée par rapport à la couverture moyenne. Au total, 46 des 52 séquences d’amorce ont pu être assignées à la référence considérée ici avec un taux d’erreur de 38,00%. La figure 6 suggère que les lectures de séquences courtes associées à la référence LC312715.1 ont également été amplifiées lors de la confirmation par PCR, malgré le fait que les séquences d’amorce n’ont pu être assignées à la référence qu’avec un taux d’erreur relativement élevé.
Enfin, passons à la référence KJ410048.1 (virus de la rougeole).
Figure 10 : Référence KJ410048.1. a) KJ410048.1_short_reads cartographié à l’aide de BBMap, (M1 ; M2) = (37 ; 0,60). b) KJ410048.1_primer cartographié à l’aide de BBMap.
La distribution de la couverture diffère sensiblement de celle de la figure 6 et présente certaines similitudes avec la distribution des lectures de séquences associées pour “rnd_uniform”, avec une variation moindre dans les zones de moindre couverture. La distribution des longueurs de lecture (Tableaux et Figures : Figure 24d)) ainsi que la longueur de lecture moyenne de 42,38 sont comparables aux données de “rnd_unifom” et sont également corrélées avec des taux d’erreur relativement élevés F1=28,70% et F2=28,79%.
Discussion et perspectives
Nous avons examiné les données de séquence publiées (numéro d’accession BioProject PRJNA603194 dans la base de données NCBI Sequence Read Archive (SRA)) sur la séquence du génome de SARS-CoV-2 (GenBank : MN908947.3) en utilisant une approche bioinformatique simple. Les méthodes que nous avons utilisées ne sont pas spécifiques au SARS-CoV-2 et peuvent être appliquées à d’autres données de séquençage sans modifications particulières.
Tout d’abord, nous avons répété la génération de contigs avec Megahit (v.1.2.9) en utilisant les données de séquence disponibles et avons obtenu des résultats significativement différents par rapport aux représentations de [1]. En particulier, nous n’avons pas été en mesure de reproduire le contig le plus long avec une longueur de 30 474 nt, qui selon [1] comprenait presque tout le génome viral et a servi de base pour la conception des amorces. Au contraire, le contig le plus long que nous avons généré (29 802 nt) a montré une correspondance presque complète avec la référence MN908947.3. Par conséquent, les données de séquence publiées ne peuvent pas être les lectures courtes originales utilisées pour la génération des contigs. Ceci est à considérer comme extrêmement problématique dans le contexte des publications scientifiques, car de cette manière il n’est plus possible de vérifier les résultats publiés. La possibilité de vérifier les hypothèses scientifiques publiées est l’essence même de la science vivante.
Contrairement à ce qui a été rapporté dans [1], il se peut que nous ayons trouvé des contigs avec une couverture élevée associés à des acides ribonucléiques (ribosomiques) d’origine humaine. Il est donc possible que tous les acides nucléiques associés à l’homme n’aient pas été éliminés lors de la construction du SARS-CoV-2. En outre, aucune preuve de la présence d’acides nucléiques viraux dans l’échantillon du patient n’a été fournie et, par conséquent, il est possible que des fragments d’acides nucléiques humains ou non viraux aient été utilisés pour construire la séquence virale revendiquée MN908947.3 dans une large mesure sans être détectés. Cette possibilité devrait être exclue par des expériences de contrôle.
Dans toutes les publications sur les génomes de référence analysés dans cette étude, les preuves nécessaires sur l’origine exacte des fragments de séquence utilisés pour la construction n’étaient pas non plus fournies et les expériences de contrôle nécessaires n’étaient pas publiées.
Nous tenons à mentionner ici que des expériences de contrôle ont peut-être déjà été réalisées de nombreuses fois sans être remarquées, ce qui montre la possibilité de construire des génomes du SARS-CoV-2 à partir d’échantillons humains non infectieux. Par exemple, le séquençage du génome entier à partir d’échantillons dont la valeur de base du Ct est supérieure à 35 est rapporté dans [5] et [17]. Cela pourrait réfuter le modèle viral du SARS-CoV-2.
L’analyse des distributions de la couverture nucléotidique ainsi que des distributions de la longueur des lectures de séquence mappables pour les séquences de référence respectives conduit à l’hypothèse d’une possible amplification involontaire de lectures de séquence non associées au SARS-CoV-2. En outre, il faut envisager la possibilité de la génération accidentelle de séquences qui n’étaient pas présentes dans l’échantillon initial mais qui ont été générées uniquement par les conditions d’amplification, telles que les séquences d’amorces utilisées et les cycles effectués. Cette possibilité nécessite donc la réalisation d’expériences de contrôle appropriées.
En plus de tenter de reproduire l’assemblage publié dans [1] avec les lectures de séquences publiées, nous avons envisagé une approche simple pour analyser la structure interne de grands ensembles de données de lectures de séquences courtes. Avec les données de séquence disponibles, nous avons pu calculer des séquences consensus pour les génomes de référence LC312715.1 (VIH) et NC_001653.2 (virus de l’hépatite delta) avec une plus grande précision que pour les séquences de référence que nous avons considérées comme associées aux coronavirus. Cela était particulièrement vrai pour la séquence bat-SL-CoVZC45 (GenBank : MG772933.1), qui a conduit à l’hypothèse d’origine du SARS-CoV-2. Ainsi, nous avons pu étayer notre hypothèse selon laquelle les séquences génomiques virales revendiquées sont des interprétations erronées dans le sens où elles ont été ou sont construites sans que cela soit remarqué à partir de fragments d’acides nucléiques non viraux. En particulier, nos résultats soulignent l’urgente nécessité de réaliser des expériences de contrôle appropriées. Pour chaque séquence génomique virale pathogène suspectée, un protocole évident serait de tenter d’assembler les séquences génomiques d’échantillons non suspectés correspondants en utilisant des protocoles identiques.
Nous avons observé des taux d’erreur R1 et R2 élevés dans les génomes de référence pour la rougeole, Ebola ou Marburg, où les fragments d’acide nucléique utilisés pour la construction ont été propagés dans des cellules Vero. La question de savoir si cela est dû aux sources d’acide nucléique elles-mêmes, aux conditions d’amplification utilisées (par exemple, les séquences d’amorces et le nombre de cycles) ou aux protocoles de séquençage (par exemple, les polymérases et les transcriptases inverses utilisées) reste ouverte.
En ce qui concerne nos résultats, outre la publication des données de séquence finales utilisées, nous recommandons toujours de publier les données de séquence résultant uniquement de l’amplification avec des hexamères aléatoires et des nombres de cycles modérés afin de fournir les données les plus impartiales possibles pour l’analyse structurelle.
Matériel et méthodes
Profondeur de couverture d’une séquence de référence avec des lectures de séquences courtes
Soit 𝐺 la longueur de la séquence de référence, Ø𝐿 la longueur moyenne de lecture, 𝑛 le nombre de lectures de séquences courtes, et 𝑁 la profondeur moyenne aléatoire de couverture de la séquence de référence avec les lectures de séquences courtes. Alors
L’expression Ø𝐿/𝐺 peut être considérée comme la probabilité de couverture d’un nucléotide dans la séquence de référence avec une lecture de séquence courte.
Génération de séquences de référence aléatoires
Le théorème suivant permet la simulation d’une variable aléatoire avec une fonction de distribution cumulative.
Théorème (principe d’inversion) [28]. Soit 𝑈 une variable aléatoire également distribuée sur l’intervalle (0,1). Soit 𝑋 une variable aléatoire avec une fonction de distribution cumulative 𝐹, et soit
Alors s’applique
Soit 𝑈𝑖,𝑖 = 1, … ,29,903 des variables aléatoires équidistantes indépendamment identiques sur l’intervalle (0,1). Soit 𝑝𝑛𝑡,𝑛𝑡 ∈{𝐴,𝑇,𝐶,𝐺} la probabilité pour le nucleotide 𝑛𝑡. Ensuite, le nucléotide 𝑁𝑖,𝑖 = 1, … ,29.903 de la séquence de référence générée de façon aléatoire est obtenu via
Pour la séquence de référence “rnd_unifom”, la distribution uniforme sur l’ensemble {𝐴,𝑇,𝐶,𝐺} a été utilisée. Pour simuler la séquence de référence aléatoire “rnd_wuhan”, l’occurrence relative des nucléotides A, T, C et G dans la séquence du génome du SARS-CoV-2 (GenBank : MN908947.3) a été choisie comme distribution des nucléotides. Dans la construction des séquences de référence randomisées “rnd_wh_mk_1” et “rnd_wh_mk_2”, la probabilité conditionnelle, respectivement sur le dernier et sur les deux derniers nucléotides, a été choisie en fonction des fréquences empiriques correspondantes dans la séquence du SARS-CoV-2 (GenBank : MN908947.3).
Simulation stochastique de couvertures aléatoires d’une séquence de référence
La fonction de distribution cumulative de la distribution exponentielle avec le paramètre 𝜆 est [28],
Soit 𝑋 une variable aléatoire avec une fonction de distribution 𝐹. Alors 𝐸𝑋 = 1/𝜆 und 𝑉𝐴𝑅𝑋 = 1/𝜆2.
3.3. Détermination de la séquence consensus finale (min. Q20)
seqtk seq -aQ64 -q20 -n N échantillon_cns.fastq > échantillon_cns.fasta
Mappage de la séquence consensus à la séquence de référence en utilisant BWA.
bwa index $reference.fasta
bwa mem $reference.fasta sample_cns.fasta > sample_cns.sam
Examen avec Tablet et Excel
L’évaluation a été réalisée à l’aide du logiciel Tablet pour la visualisation des données de séquence et du programme de feuille de calcul Excel.
Références
[1] Fan Wu u. a. A new coronavirus associated with human respiratory disease in China. In: Nature 580.7803 (2020). DOI: 10.1038/s41586-020-2202-3.
[2]Na Zhu u. a. A Novel Coronavirus from Patients with Pneumonia in China, 2019. In: New England Journal of Medicine 382.8 (2020), S. 727-733. DOI:10.1056/nejmoa2001017.
[3]Divinlal Harilal u. a. SARS-CoV-2 Whole Genome Amplication and Sequencing for Effective Population-Based Surveillance and Control of Viral Transmission. In: Clinical Chemistry 66.11 (2020), S. 1450-1458. DOI: 10.1093/clinchem/hvaa187.
[4]Jalees A. Nasir u. a. A Comparison of Whole Genome Sequencing of SARSCoV-2 Using Amplicon-Based Sequencing, Random Hexamers, and Bait Capture. In: Viruses 12.8 (2020), S. 895. DOI: 10.3390/v12080895.
[5]Clinton R. Paden u. a. Rapid, sensitive, full-genome sequencing of severe acute respiratory syndrome coronavirus 2. In: Emerging Infectious Diseases 26.10 (2020), S. 2401-2405. DOI: 10.3201/eid2610.201800.
[6]Sureshnee Pillay u. a. Whole Genome Sequencing of SARS-CoV-2: Adapting Illumina Protocols for Quick and Accurate Outbreak Investigation during a Pandemic. In: Genes 11.8 (2020), S. 949. DOI: 10.3390/genes11080949.
[7]Dan Hu u. a. Genomic characterization and infectivity of a novel SARS-like coronavirus in Chinese bats. In: Emerging Microbes & Infections 7.1 (2018), S. 1-10. DOI: 10.1038/s41426-018-0155-5.
[8] Davaalkham Jagdagsuren u. a. The second molecular epidemiological study of HIV infection in Mongolia between 2010 and 2016. In: Plos One 12.12 (2017). DOI: 10.1371/journal.pone.0189605.
[9] J. A. Saldanha, H. C. Thomas und J. P. Monjardino. Cloning and sequencing of RNA of hepatitis delta virus isolated from human serum. In: Journal of General Virology 71.7 (1990), S. 1603-1606. DOI: 10.1099/0022-1317-71-7-1603.
[10] Jernej Mlakar u. a. Zika Virus Associated with Microcephaly. In: New EnglandJournal of Medicine 374.10 (2016), S. 951-958. DOI: 10.1056 /nejmoa1600651.
[11] Christopher L. Parks u. a. Comparison of Predicted Amino Acid Sequences of Measles Virus Strains in the Edmonston Vaccine Lineage. In: Journal of Virology 75.2 (2001), S. 910-920. DOI: 10.1128/jvi.75.2.910-920.2001.
[12] Konstantin M. J. Sparrer u. a. Complete Genome Sequence of a Wild-Type Measles Virus Isolated during the Spring 2013 Epidemic in Germany. In: Genome Announcements 2.2 (2014). DOI: 10.1128/genomea.00157-14.
[13] Paul A. Rota u. a. Characterization of a Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. In: Science 300.5624 (2003), S. 1394-1399. DOI: 10.1126/science.1085952.
[14] Runtao He u. a. Analysis of multimerization of the SARS coronavirus nucleocapsid protein. In: Biochemical and Biophysical Research Communications 316.2 (2004), S. 476-483. DOI: 10.1016/j.bbrc.2004.02.074.
[15] Tracey Goldstein u. a. The discovery of Bombali virus adds further support for bats as hosts of ebolaviruses. In: Nature Microbiology 3.10 (2018), S. 1084-1089. DOI: 10.1038/s41564-018-0227-2.
[16] Jonathan S. Towner u. a. Marburgvirus Genomics and Association with a Large Hemorrhagic Fever Outbreak in Angola. In: Journal of Virology 80.13 (2006), S. 6497-6516. DOI: 10.1128/jvi.00069-06.
[17] Annika Brinkmann u. a. Amplicov: Rapid whole-genome sequencing using multiplex PCR amplication and real-time Oxford Nanopore minion sequencing enables rapid variant identication of SARS-COV-2. In: Frontiers in Microbiology 12 (2021). DOI: 10.3389/fmicb.2021.651151.
[20a] Dinghua Li u. a. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. In: Bioinformatics 31.10 (2015), S. 1674-1676. DOI: 10.1093/bioinformatics/btv033.
[21a] Shifu Chen u. a. fastp: an ultra-fast all-in-one FASTQ preprocessor. In: Bioinformatics 34.17 (2018), S. i884-i890. DOI: 10.1093/bioinformatics/bty560.
[21b] OpenGene. OpenGene/fastp: An ultra-fast all-in-one FASTQ preprocessor(QC/adapters/trimming/ltering/splitting/merging…) URL: https://github.com/OpenGene/fastp.
[22a] Ben Langmead u. a. Scaling read aligners to hundreds of threads on generalpurpose processors. In: Bioinformatics 35.3 (2018), S. 421-432. DOI: 10. 1093/bioinformatics/bty648.
[27a] Wei Shen u. a. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. In: Plos One 11.10 (2016). DOI: 10.1371/journal.pone.0163962.
TABLEAU DE COMPARAISON ENTRE LES LAITS CRUS ET PASTEURISÉS
Catégorie comparée
Lait cru
Lait pasteurisé
1) Enzymes
Toutes disponibles.
Moins de 10% restantes.
2) Protéines :
100% disponibles, les 22 acides aminés, dont 8 essentiels.
Les protéines-lysine et tyrosine sont altérées par la chaleur avec une grave perte de disponibilité métabolique. Cela a pour conséquence de rendre l’ensemble du complexe protéique moins disponible pour la réparation et la reconstruction des tissus.
3) Les graisses : (des études indiquent que les graisses sont nécessaires pour métaboliser les protéines et le calcium. Tous les aliments naturels contenant des protéines contiennent des graisses).
Les 18 acides gras métaboliquement disponibles, tant les graisses saturées que les graisses insaturées.
Altérée par la chaleur, notamment les 10 graisses insaturées essentielles.
4) Vitamines :
Toutes 100% disponibles.
Parmi les vitamines liposolubles, certaines sont classées comme instables et une perte est donc provoquée par le chauffage au-dessus de la température du sang. Cette perte de vitamines A, D, E et F peut atteindre 66 % et celle de la vitamine C dépasse généralement 50 %. Les pertes de vitamines hydrosolubles sont affectées par la chaleur et peuvent aller de 38 % à 80 %.
5) Glucides :
Facilement utilisables dans le métabolisme. Toujours associés naturellement aux éléments.
Les tests indiquent que la chaleur a apporté quelques modifications rendant les éléments moins disponibles métaboliquement.
6) Les minéraux :
Tous 100% métaboliquement disponibles. Les principaux composants minéraux sont le calcium, le chlore, le magnésium, le phosphore, le potassium, le sodium et le soufre. Les oligo-éléments vitaux, au nombre de 24 ou plus, sont tous disponibles à 100 %.
Le calcium est altéré par la chaleur et la perte dans le métabolisme peut atteindre 50 % ou plus, selon la température de pasteurisation. Perte d’autres minéraux essentiels, car un minéral agit généralement en synergie avec un autre élément. Il y a une perte d’enzymes qui servent de conducteurs dans l’assimilation des minéraux.
NOTE :
La croissance bactérienne dans le lait cru augmente très lentement, car les bactéries acidifiantes amicales (l’antiseptique de la nature) retardent la croissance des organismes envahisseurs (les bactéries). Il se conserve généralement pendant plusieurs semaines lorsqu’il est placé au réfrigérateur et tourne au lieu de pourrir.
La pasteurisation est le processus qui consiste à chauffer chaque particule de lait à une température d’au moins 63°c et à le maintenir à cette température pendant au moins 15 secondes. La pasteurisation n’élimine pas la saleté, ni les toxines produites par les bactéries du lait. La croissance bactérienne sera géométriquement rapide après la pasteurisation et l’homogénéisation. Le lait devient progressivement rance en quelques jours, puis se décompose.
En 1945, 450 cas de maladies infectieuses ont été attribués au lait cru. Il y a eu 1 492 cas attribués au lait pasteurisé.[1] Il y a eu 1 cas de maladie pour 12 400 000 litres de lait pasteurisé consommés, et 1 cas de maladie pour 18 900 000 litres de lait cru consommés.[2] En d’autres termes, une personne pouvait boire 6 500 000 litres de lait cru de plus que de lait pasteurisé sans tomber malade.
En 1945, une épidémie d’intoxication alimentaire s’est produite à Phoenix, en Arizona.[3] Le rapport officiel indique que ” les relevés de pasteurisation… montrent que le lait a été correctement pasteurisé et permettent de supposer que la toxine a été produite dans le lait pendant son stockage… ” Trois cents (300) personnes ont été malades à la suite de cet incident d’intoxication alimentaire au lait pasteurisé.
Great Bend, Kansas, en 1945, a signalé 468 cas de gastro-entérite dus au lait pasteurisé. Ces cas ont été attribués à “des conditions insalubres dans les laiteries, des bouteilles non stérilisées”. 9 personnes sont décédées.
En octobre 1978, une épidémie de salmonelle a été attribuée à un empoisonnement alimentaire par du lait pasteurisé, impliquant 68 personnes en Arizona. Le taux de bactéries était 23 fois supérieur à la limite légale. Le CDC a signalé que le lait avait été correctement pasteurisé, mais il continue d’insister sur le fait que ” seule la pasteurisation offre une garantie contre les infections “.
En juin 1982, 172 personnes dans une région de trois États du Sud-Est ont été frappées par une infection intestinale. Plus de 100 ont été hospitalisées. L’infection, qui a provoqué une diarrhée sévère, de la fièvre, des nausées, des douleurs abdominales et des maux de tête, a été attribuée au lait pasteurisé[4].
En 1983, lors d’une épidémie de listériose au Massachusetts, le lait pasteurisé entier ou à 2 % a été impliqué comme source d’infection. L’inspection de l’usine de production de lait n’a détecté aucune défaillance apparente dans le processus de pasteurisation.[5]
En août 1984, environ 200 personnes ont été infectées par S. typhimurium à partir de lait pasteurisé produit dans une usine de Melrose Park, IL. Les autorités de réglementation ont gardé cette épidémie secrète. Sans preuve, ils ont conclu que le lait n’avait pas été correctement pasteurisé. Mais, de nouveau, en novembre 1984, une autre épidémie de S. typhimurium s’est déclarée chez des personnes ayant consommé du lait pasteurisé mis en bouteille dans la même usine. Encore une fois, ils ont gardé le secret et ont supposé que le lait n’avait pas été correctement pasteurisé. Puis, en mars 1985, il y a eu 16 284 cas confirmés de S. typhimurium résultant du lait pasteurisé mis en bouteille dans la même usine. Les tests ont prouvé que le lait avait été correctement pasteurisé. Des enquêteurs ayant des idées préconçues selon lesquelles le lait n’avait pas été correctement pasteurisé, alimentés par les efforts des services de santé, ont tiré des conclusions sans enquête et ont accusé le lait cru. Les médias ont à leur tour relayé cette théorie auprès du public.[6]
Consumer Reports, janvier 1974, a révélé que sur 125 échantillons testés de lait et de produits laitiers pasteurisés, 44% se sont révélés en violation des réglementations de l’état. Consumer Reports a conclu que “la qualité d’un certain nombre de produits laitiers dans cette étude était tout simplement déplorable”. Consumer Reports a déclaré que les “anciennes objections” au lait pasteurisé sont toujours valables aujourd’hui :
a) La pasteurisation est une excuse pour la vente de lait sale. b) La pasteurisation peut être utilisée pour masquer un lait de mauvaise qualité. c) La pasteurisation encourage la négligence et décourage les efforts pour produire du lait propre.
L’Union des consommateurs a rapporté en juin 1982 que des bactéries coliformes ont été trouvées dans de nombreux échantillons testés de produits laitiers pasteurisés. Certains comptages atteignaient 2200 organismes par centimètre cube.
Exemples d’épidémies attribuées à des intoxications alimentaires bactériennes dues au lait pasteurisé :
1945 : 1 492 cas pour l’année aux États-Unis.
1945 : 1 foyer, 300 cas à Phoenix, Arizona.
1945 : Plusieurs foyers, 468 cas de gastro-entérite, 9 décès, à Great Bend, Kansas.
1978 : 1 foyer, 68 cas en Arizona.
1982 : Plus de 17 000 cas d’entérocolite à Yersinia à Memphis, Tennessee.
1982 : 172 cas, dont plus de 100 hospitalisés, dans une région de trois États du Sud.
1983 : 1 foyer, 49 cas de listériose dans le Massachusetts.
1984 : Août, 1 foyer de S. typhimurium, environ 200 cas, dans une usine à Melrose Park, IL.
1984 : Novembre, 1 foyer de S. typhimurium, dans la même usine de Melrose Park, IL.
Mars : 1985, 1 foyer, 16 284 cas confirmés, dans la même usine de Melrose Park, IL.
1985 : 197 000 cas d’infections à Salmonella résistantes aux antimicrobiens dans une laiterie de Californie.[7]
1985 : Plus de 1 500 cas, culture de Salmonella confirmée, dans le nord de l’Illinois.
1993 : 2 épidémies dans tout l’État, 28 cas d’infection à Salmonella.
1994 : 3 épidémies, 105 cas, E. Coli et Listeria en Californie.
1995 : 1 foyer, 3 cas en Californie.
1996 : 2 foyers Campylobactor et Salmonella, 48 cas en Californie.
1997 : 2 épidémies, 28 cas Salmonella en Californie.
Le professeur Fosgate, du département des sciences laitières de l’université de Géorgie, a déclaré : “La pasteurisation a été prêchée comme une garantie à cent pour cent pour le lait. Ce n’est tout simplement pas vrai. Si le lait est contaminé aujourd’hui, il y a de fortes chances que ce soit après la pasteurisation.”
Le Rapport Flexner est la clé qui a permis à la famille Rockefeller, déjà milliardaire grâce au pétrole et à la famille Carnegie, milliardaire via l’acier et le business du chemin de fer, d’instaurer la médecine moderne, celle qui allait promouvoir “la science” des Labos pharmaceutiques.
C’est lors de l’Info en QuestionS #84, le 20.01.22, que j’ai évoqué ce fameux rapport qui a permis de fermer 117 des 148 écoles de médecine du début du siècle dernier aux Etats-Unis et de reléguer les médecines holistique naturelles au rang de “sectes”.
Pour déterminer la validité d’une théorie scientifique touchant une discipline particulière, il est bien davantage utile de connaître la démarche scientifique et le propre de la science que d’être un expert du domaine en question. Le propre de la science est le doute, la remise en question des connaissances acquises à la lumière de nouvelles données ou informations, par exemple suite à l’exploration de nouvelles voies ou à l’observation du réel sous un nouvel angle de vue ou selon une nouvelle perspective. Une théorie explicative de phénomènes observés ne peut être véritablement d’ordre scientifique que si elle est contestable par l’argumentation ou l’expérimentation dans le cadre de la démarche scientifique. Si a contrario elle se pose en vérité absolue ou définitive, alors elle ne relève pas de science, mais de superstition ou de croyances religieusement ou affectivement entretenues. À la lumière de nombreux faits et constats, nous allons tenter de montrer ici en quoi la théorie virale ne relève pas de science, mais de scientisme et de croyances erronées.
1)- La démarche scientifique
Il s’agit d’une méthode rigoureuse pour tenter d’expliquer la réalité objective, présentée schématiquement ici.
Les étapes de la démarche ou de la méthode scientifique
1.L’analyse
Elle consiste à subdiviser l’étude en éléments plus simples aux propriétés et fonctions particulières.
La modélisation
La modélisation est une phase de l’analyse consistant à rechercher les interactions entre les éléments de l’objet d’étude, dont les relations de cause à effet qui peuvent exister entre eux ou vis-à-vis de systèmes extérieurs.
La démonstration d’un lien de causalité
De manière rigoureuse, pour établir l’existence d’une relation de cause à effet entre un phénomène ou élément A et un phénomène ou élément B, il est nécessaire et suffisant de vérifier les conditions exposées ci-après.
La démonstration scientifique s’appuie sur la logique. En logique pure, pour qu’un phénomène, un événement, un objet ou un être vivant soit la cause unique d’un autre phénomène, événement, objet ou être vivant, il doit respecter les conditions logiques suivantes :
Il est suffisant que la présence de la cause entraîne la présence de la conséquence prévue ou inversement que l’absence de la conséquence implique l’absence de la cause.
Si la cause est effectivement unique, il n’y a bien sûr aucune autre cause systématiquement présente.
Il est par contre nécessaire que l’absence de la cause entraîne l’absence de la conséquence prévue, ou qu’inversement la présence du phénomène prévu implique la présence de la cause.
Exemple en climatologie :
Si les nuages étaient la cause unique de la pluie, il serait suffisant que la présence de nuages entraîne celle de la pluie et inversement l’absence de pluie impliquerait l’absence de nuages.
Or, on observe assez souvent la présence de nuages sans qu’il y ait de pluie du tout et dans certains cas, au lieu de pluie, on a plutôt de la neige ou de la grêle.
De plus, il serait nécessaire que l’absence de nuages entraîne l’absence de la pluie ou inversement que la présence de la pluie implique celle des nuages.
Mais dans la pratique, on a pu pourtant observer de la pluie fine en certains lieux et circonstances sans nuages visibles, par phénomène de condensation localisée de vapeur d’eau.
Donc, tout cela montre logiquement que les nuages ne sont pas la cause unique de la pluie et que d’autres facteurs (ou éléments causaux) interviennent, comme la température et la pression de l’atmosphère.
Exemple en virologie :
Si un virus donné (comme le supposé SRAS-CoV-2 pourtant jamais physiquement isolé) était la cause unique de la maladie Covid-19 qu’on lui attribue, alors il serait suffisant que sa présence entraîne la maladie associée, et inversement l’absence de la maladie impliquerait l’absence du virus.
Or, on observe assez souvent que des personnes auraient prétendument le SRAS-CoV-2 dans leur organisme alors qu’elles ne sont pas malades et n’ont aucun symptôme, au point qu’on les appelle « malades asymptomatiques ».
De plus, il serait nécessaire que l’absence du virus entraîne l’absence de la maladie ou qu’inversement la présence de la maladie entraîne celle du virus.
Or, comme les symptômes de cette maladie couvrent d’autres maladies ou phénomènes (comme les symptômes de la grippe ou comme les effets sur la santé d’ondes électromagnétiques pulsées) et que l’on a pu les observer sans le moindre lien avec le SRAS-CoV-2, on peut dire que l’on a des personnes qui aujourd’hui seraient diagnostiquées Covid-19 sans pour autant que l’on y détecte de SRAS-CoV-2.
Donc tout cela montre qu’au mieux le SRAS-CoV-2 n’est pas la cause unique de la Covid-19, mais que d’autres facteurs doivent logiquement intervenir s’il a quelque chose à y voir (et si par hasard il existe vraiment, mais n’est pas un simple artefact virtuel produit d’une méthodologie qui n’a rien de scientifique).
Exemple en électricité :
Si le courant électrique est la cause unique de la lumière d’une lampe, il est suffisant que la présence du courant dans le filament de l’ampoule entraîne son éclairage et inversement, l’absence de courant implique l’absence de lumière.
Ceci est effectivement observé à partir du moment où le courant est d’intensité suffisante. (En toute rigueur, on pourrait toutefois ici invoquer une cause secondaire, à savoir l’existence d’une résistance électrique au passage du courant dans le filament. Et donc en toute rigueur le courant électrique est la cause principale, mais pas unique de l’éclairage de la lampe.)
De plus, il est nécessaire que l’absence de courant entraîne l’absence de lumière dans l’ampoule ou inversement que la présence de lumière implique celle du courant.
On n’observe effectivement pas de lampes électriques allumées sans qu’elles soient parcourues par un courant électrique
2. L’épreuve des faits
Ce n’est pas parce qu’un raisonnement est juste qu’il correspond aux faits objectifs. L’analyse intellectuelle d’une situation peut sembler cohérente et logique de prime abord, mais ne pas pour autant avoir le moindre rapport avec le réel.
Un raisonnement fallacieux qui n’a que l’apparence de la logique est appelé un « sophisme ».
Un raisonnement dissocié de la réalité est nommé « syllogisme ».
Exemple :
Le raisonnement suivant relève à la fois de syllogisme et de sophisme :
Tout ce qui est rare est cher ;
Les chevaux bon marché sont rares ;
donc les chevaux bon marché sont chers.
Il est sophiste, car la conclusion est en contradiction interne et se trouve donc fausse.
Il relève de syllogisme, car en réalité les chevaux bon marché sont peu chers, par définition et constat, et aussi parce que tout ce qui est rare n’est pas nécessairement cher, comme certaines formes de bactéries, de moisissures, d’insectes, ou encore de « mauvaises herbes » que pourtant personne n’aurait l’idée d’acheter et dont la valeur marchande est alors très voisine de zéro (quelle que soit l’unité monétaire considérée).
Les mesures
L’épreuve des faits consiste en particulier à effectuer des expériences, des mesures et des évaluations pour vérifier leur conformité à ce que prévoit la théorie. Une bonne théorie doit être prédictive.
Pour être significatifs, les expériences, les tests et les essais doivent être reproduits autant de fois que nécessaire afin de réduire la part d’erreurs et des biais psychologiques (comme ceux provenant de conflits d’intérêt ou de corruption et qui peuvent amener à fausser les résultats).
Contre-expériences
Appelées aussi expériences de contrôle ou encore expériences témoins, elles consistent à éliminer au moins une des causes supposées être responsables d’un des phénomènes étudiés dans le cadre de la théorie pour tenter pourtant d’obtenir les mêmes effets.
Par exemple, pour valider la théorie virale, il ne suffit pas d’observer de multiples fois des tissus infectés par un virus donné, puis de constater la mort des cellules biologiques pour conclure que le virus est bien le responsable de la maladie concernée. Il faut aussi réaliser plusieurs fois des contre-expériences dans lesquelles on a recours à exactement les mêmes conditions expérimentales à l’exception de la présence du virus. Si malgré l’absence de ce dernier les cellules meurent similairement, cela démontre que le virus n’en était pas la cause.
3. Retour à l’analyse
Si les mesures, les expériences et/ou les contre-expériences ne correspondent pas à la théorie ou ne parviennent pas à la valider, il est nécessaire de l’abandonner ou au moins de la revoir plus ou moins complètement.
2)- La théorie virale
Cette théorie suppose depuis 1954 que des virus de taille nettement plus petite que celle des bactéries et des cellules végétales, animales ou humaines sont susceptibles d’y pénétrer pour s’y multiplier en utilisant leur matériel génétique, puis d’en sortir pour aller infecter d’autres individus de même espèce, voire d’autres espèces, alors qu’ils ne sont pas eux-mêmes vivants. L’effet d’une telle invasion parasitaire serait alors l’apparition subséquente des maladies non bactériennes telles que la grippe, la variole et la polio. Le lien de causalité supposé est le suivant : la cause est le virus et l’effet automatique est alors une maladie virale spécifique aux symptômes caractéristiques. Le gros problème est que l’existence d’un tel lien causal n’a jamais été établie pour aucun des supposés virus, comme nous allons le voir, et que l’existence même de telles particules (non vivantes qui seraient externes aux organismes vivants végétaux, animaux et humains, mais guère capables de survivre hors d’eux plus de quelques heures) n’est pas non plus scientifiquement et rigoureusement démontrée.
Que font les virologues lorsqu’ils prétendent isoler un virus ?
« Les virologistes croient aux virus parce qu’ils ajoutent au tissu et aux cultures cellulaires du sang, de la salive ou d’autres fluides corporels supposés infectés — après avoir retiré les nutriments de la culture cellulaire respective et après avoir commencé à l’empoisonner avec des antibiotiques toxiques. Ils pensent que la culture cellulaire est alors tuée par les virus » ~ Dr Stefan Lanka, qui ne revendique plus le titre de « virologue », dans https://nouveau-monde.ca/la-fausse-idee-appelee-virus-la-rougeole-a-titre-dexemple
« Les virologues prélèvent des échantillons non purifiés sur un nombre relativement restreint de personnes, souvent moins de dix, atteintes d’une maladie similaire. Ils procèdent ensuite à un traitement minimal de cet échantillon et l’inoculent à une culture tissulaire contenant habituellement quatre à six autres types de matériel, qui contiennent tous du matériel génétique identique à ce que l’on appelle un « virus ». La culture de tissus est affamée et empoisonnée et se désintègre naturellement en de nombreux types de particules, dont certaines contiennent du matériel génétique. À l’encontre de tout bon sens, de toute logique, de l’usage de la langue anglaise et de l’intégrité scientifique, ce processus est appelé « isolement du virus. » Cette infusion [ou soupe chimico-génétique] contenant des fragments de matériel génétique provenant de nombreuses sources est ensuite soumise à une analyse génétique, qui crée alors dans un processus de simulation informatique la séquence présumée du prétendu virus, un génome dit « in silico » [du mot « silicium », pour indiquer sa nature purement logicielle, puisque les microprocesseurs à la base du fonctionnement des ordinateurs sont faits de semi-conducteurs en silicium]. À aucun moment un virus réel n’est confirmé par microscopie électronique. À aucun moment, un génome n’est extrait et séquencé à partir d’un virus réel » ~ Dr Andrew Kaufman, dans https://nouveau-monde.ca/declaration-sur-lisolement-des-virus
« La théorie des virus pathogènes remonte au XIXe siècle et les virologues ont passé la première moitié du XXe siècle à essayer d’extraire ces virus présumés directement d’hôtes vivants. Les échecs répétés les ont amenés à changer de cap dans les années 1950 afin de conserver une quelconque crédibilité. Les virologues devaient fournir quelque chose à montrer à leurs investisseurs potentiels, y compris à l’industrie pharmaceutique en plein essor qui s’impatientait de développer des vaccins et des traitements antimicrobiens.
En 1954, des scientifiques ont rapporté qu’ils avaient des preuves de la présence du virus de la rougeole en se basant sur l’observation qu’un échantillon provenant d’un patient atteint de la rougeole avait tué certaines cellules dans un tube à essai. Ces phénomènes sont connus sous le nom d’”effets cytopathiques”. Les auteurs ont admis que “bien qu’il n’y ait aucune raison de conclure que les facteurs in vivo [chez l’homme] sont les mêmes que ceux qui sous-tendent la formation de cellules géantes et les perturbations nucléaires in vitro [dans le tube à essai], l’apparition de ces phénomènes… pourrait être associée au virus de la rougeole”.
L’apparition du CPE (effet cytopathique) est à la base des allégations frauduleuses d’isolement et de pathogénicité de la virologie moderne : un échantillon (par exemple, un écouvillon nasal) est prélevé sur un patient et mélangé à des cellules dans un tube à essai, les cellules meurent et l’on déclare qu’un virus a été “isolé”. Ce que les virologues ne veulent pas que vous sachiez, c’est que les mêmes résultats peuvent être obtenus sans ajouter de prétendus échantillons de virus dans le tube à essai — en d’autres termes, c’est le processus lui-même, la privation de nourriture de la cellule et l’ajout de diverses substances toxiques telles que des antibiotiques et des antifongiques, qui provoquent la réaction et la mort des lignées cellulaires déjà anormales, sans qu’aucun virus ne soit nécessaire. (Des photographies d’infections “factices” sont parfois fournies, mais les détails de ces expériences se distinguent par leur absence).
Il y a, bien sûr, les images de ce qu’on prétend être le virus à l’origine de tous nos problèmes. Cependant, ces images 3D colorées ne sont rien d’autre que des images générées par ordinateur constituant une représentation artistique » ~ Docteurs Mark Bailey et John Bevan-Smith dans https://nouveau-monde.ca/la-fraude-du-covid-19-et-la-guerre-contre-lhumanite
Pour simplifier, les étapes du processus qui prétend isoler un virus et le caractériser sont les suivantes :
récolte de matériel biologique infecté (sang, morve, peau, etc. selon le cas) ;
prétendue « culture » des virus dans une préparation qui mélange le matériel infecté, des cellules prétendument propices à la culture virale, des antibiotiques et des antifongiques toxiques et des substances susceptibles de favoriser la multiplication virale ;
après la mort des cellules de culture, centrifugation de la soupe résultante pour séparer les composants par densité et recueillir la couche supposée contenir les virus reproduits ;
séquençage par logiciel informatique qui découpe d’abord en morceaux le matériel génétique recueilli avant de reconstituer virtuellement la supposée séquence génétique complète du supposé virus d’après un modèle de base supposé être celui du virus ou très proche de lui.
3)- Confrontation de la théorie virale à la rigueur scientifique
Avant même de chercher à déterminer la réalité ou non d’un lien causal entre virus et maladies qualifiées de virales, si au moins l’une des quatre étapes qui viennent d’être évoquées est démontrée sans fondement scientifique, alors c’est déjà suffisant pour faire s’effondrer la théorie puisqu’alors cela confirmerait qu’aucun virus ne peut être véritablement isolé en suivant une telle procédure. Et s’il ne peut pas être isolé, il devient impossible de démontrer ensuite qu’il provoque bien une maladie.
À notre connaissance, un seul scientifique s’est jusqu’à maintenant donné la peine d’effectuer des expériences de contrôle pour vérifier ou pour infirmer la validité du supposé processus d’isolement, de purification et de caractérisation d’un virus donné. Il s’agit de Stefan Lanka (Voir en particulier :
Lors de la première phase, en plus des cellules humaines récoltées, on trouve au minimum le matériel génétique des cellules de cultures, mais aussi possiblement des cellules de chien, de chat ou de quoi que soit d’autre qui se trouve dans l’environnement de l’individu malade ou supposément infecté d’un virus.
Pour ce qui concerne la seconde phase, Lanka a effectué des expériences témoin qui démontrent que l’on parvient à faire mourir les cellules de culture sans ajouter aucun tissu biologique infecté, mais par le seul fait du processus lui-même. En d’autres termes, il a démontré ainsi que ce sont les conditions expérimentales seules qui produisent l’effet de mort cellulaire et de supposée multiplication virale qui en découle, ceci à cause des produits chimiques toxiques ajoutés et de l’insuffisance ou de l’inadéquation des nutriments reçus par les cellules de culture.
Lors de la troisième phase, en raison de la mixture présente dans l’étape 1, et compte tenu de l’absence à ce stade d’expériences de contrôle, il n’y a aucune assurance que le matériel génétique récolté après centrifugation et purification correspond uniquement à celui du malade ou de la personne soi-disant viralement infectée.
Et concernant la dernière phase, Lanka a démontré qu’il pouvait obtenir n’importe quelle séquence génétique virale, puisqu’au cours du processus, on commence par découper les brins d’ADN en petites séquences de nucléotides avant de les soumettre à un complexe processus de reconstitution logicielle lors duquel on élimine des éléments indésirables qui ne cadrent pas avec le modèle viral supposé et pour lequel on comble les trous en fonction de ce modèle, ceci à volonté. De plus et au final et le plus souvent, le puzzle ainsi reconstitué ne correspond même pas à 100 % du génome supposé. Pour donner une idée de la « valeur » scientifique d’une telle procédure qui n’est nullement expérimentale, mais purement informatique, ce procédé est similaire à celui qui consisterait à découper en petits morceaux les feuilles d’un millier de romans, à les mélanger au hasard, puis à s’efforcer de reconstituer un roman donné (par exemple Les quatre mousquetaires) sans même être certain que ce roman faisait partie du lot des romans découpés en morceaux. Si les morceaux de feuilles sont suffisamment petits, après moult ajustements, rabotage de mots dans certains bouts de phrases, ou ajout de termes dans d’autres morceaux, on pourra presque sûrement à un moment donné s’approcher du roman voulu, disons avec le même pourcentage de précision que pour la séquençage génétique, à savoir quelque chose comme 99,7%. Si le pourcentage descend trop bas, on parlera alors d’un roman « variant » : Omicron ou Delta, ou quoi que ce soit d’autre. Et si on est plus proche d’un des variants déjà trouvés que du roman original, on dira alors qu’on a affaire à un sous-variant. Quoi qu’il en soit, on ne reconstitue ainsi quasiment jamais le roman cherché et d’autre part, rien ne permet de s’assurer que ce roman viral était bien présent dans la mixture initiale.
Aucune des quatre étapes n’est scientifiquement rigoureuse, et lorsque Lanka a effectué les expériences témoins indispensables pour une telle rigueur, il a tout simplement démontré qu’il pouvait obtenir tout et n’importe quoi en matière de prétendus virus. Lui et quelques autres chercheurs ou médecins se sont penchés sur la littérature existante en matière d’isolement de virus et ont pu constater qu’aucun texte ne démontre scientifiquement et rigoureusement l’existence d’un virus avec un code génétique bien déterminé et qui aurait pu être simultanément observé par microscopie. La totalité du processus relève essentiellement de scientisme, voire de fraude involontaire ou intentionnelle, mais ne démontre scientifiquement rien et surtout pas l’existence de virus pathogènes non vivants, mais parasites qui se multiplieraient dans des cellules vivantes.
« La mort des tissus/cellules est également considérée comme l’isolation d’un virus parce qu’ils prétendent que quelque chose de l’extérieur, d’un autre organisme, a été apporté dans le laboratoire, bien qu’un virus n’ait jamais été isolé au sens du mot isolation, et n’ait jamais été photographié et caractérisé biochimiquement en tant que structure unique entière. Les micrographies électroniques des supposés virus montrent en réalité des particules cellulaires assez normales provenant de tissus et cellules en train de mourir, et la plupart des photos ne montrent qu’un modèle informatique (CGI — computer generated images — images générées par ordinateur ou images de synthèse — NdT). Parce que les parties impliquées CROIENT également que les tissus et cellules en train de mourir deviennent eux-mêmes des virus, leur mort est également considérée comme la propagation du virus. Les parties impliquées croient toujours ceci parce que le découvreur de cette méthode a reçu le Prix Nobel et ses articles sur les « virus » restent des articles de référence » ~ Stefan Lanka dans https://nouveau-monde.ca/la-fausse-idee-appelee-virus-la-rougeole-a-titre-dexemple
4)- Véritable cause des maladies et des apparentes contagions
La théorie virale s’avérant bidon, Lanka et quelques autres pionniers ont redécouvert les anciennes théories abandonnées à cause d’intérêts pharmaceutiques puissants et les ont améliorées à la lumière des dernières découvertes, notamment celle de l’existence scientifiquement démontrée elle des exosomes. Ces composants sont produits par les cellules organiques lorsqu’elles sont agressées d’une manière ou d’une autre par des toxines, par le froid, par des ondes électromagnétiques, par des champs électriques, par des émotions négatives ou par d’autres facteurs stressants. Ils ont la même apparence que celle attribuée à certains modèles de virus, parce que notamment le scientisme moderne de base matérialiste tend trop souvent à inverser causes et effets.
« Toutes les affirmations qui disent que les virus sont des agents pathogènes sont fausses et sont basées sur des erreurs d’interprétation facilement identifiables, compréhensibles et vérifiables. Les vraies causes des maladies et phénomènes qui sont imputées aux virus ont déjà été découvertes et recherchées : cette connaissance est désormais disponible. Tous les scientifiques qui pensent qu’ils travaillent en laboratoire sur des virus sont en fait en train de travailler sur des particules habituelles de tissus spécifiques en train de mourir ou sur des cellules qui ont été préparées d’une manière spéciale. Ils croient que ces tissus et cellules sont en train de mourir parce qu’ils ont été infectés par un virus. En réalité, les cellules et tissus infectés étaient en train de mourir parce qu’ils étaient affamés et empoisonnés suite aux expériences dans le laboratoire » ~ Stefan Lanka dans https://nouveau-monde.ca/la-fausse-idee-appelee-virus-la-rougeole-a-titre-dexemple
Par ailleurs, des expériences assez récentes tendent à démontrer que le corps humain, de nature électrique et magnétique, est également sensible à distance à la condition vitale des autres. Les phénomènes physiques de résonance et d’induction magnétique peuvent en partie expliquer l’influence contagieuse de la bonne ou au contraire de la mauvaise santé des uns sur les autres, en plus du fait que bien des prétendues « épidémies » ne résultent le plus souvent que de l’exposition à des facteurs communs à une région donnée, voire dans certains cas à la planète entière. Et dans pratiquement tous les cas d’épidémies historiques, on a pu trouver des explications alternatives à celle de bactéries ou de virus.
Dans le cas particulier de la maladie Covid-19 qui regroupe de multiples symptômes (grippaux, cardio-vasculaires, etc.), il se trouve que ces symptômes ont été démontrés comme pouvant être causés par diverses ondes électromagnétiques, surtout sous forme artificielle pulsée et particulièrement dans la bande des micro-ondes. C’est comme par hasard le cas des ondes Wi-Fi, 3G, 4G, 4G+ et 5G. Et il apparaît que les lieux d’apparition de la Covid coïncident temporellement et géographiquement de manière plutôt précise avec celle de l’implantation de la 5G à laquelle le corps humain n’a pas encore eu le temps de s’adapter. Il apparaît également que des facteurs additionnels tels que la quantité de métaux, notamment lourds, dans le corps sont susceptibles d’aggraver les symptômes, car alors les cellules du corps éprouvent davantage de difficulté pour se purifier de l’agression subie. Le corps humain est de nature électrique et magnétique très sensible et l’existence des électrocardiogrammes et des électroencéphalogrammes en est une des preuves. Les ondes électromagnétiques pulsées évoquées ci-dessus sont fortement susceptibles de perturber les délicats équilibres et mécanismes biologiques qui reposent sur cette nature électrique et magnétique, comme le cycle énergétique de Krebs, les influx nerveux et les processus neuronaux. Chercher à ridiculiser la probable ou au moins possible causalité entre 5G et Covid ne relève pas de science, mais d’obscurantisme scientiste et de fermeture d’esprit ou au mieux de soumission à des intérêts financiers liés à l’industrie pharmaceutique et/ou à celle des télécommunications, secteurs particulièrement influents de nos jours.
Alain Scohy, né en 1947 à Talence en Gironde, est docteur en médecine, diplômé de la Faculté de Médecine de Montpellier en 1973.
Alain Scohy a tout d’abord commencé sa carrière en tant que Médecin de campagne dans l’Aveyron pendant cinq ans. Dans le cadre de sa pratique, il est très rapidement effaré par la santé déplorable des petits enfants. Il mettra trois ans à en comprendre l’une des causes : la pratique des vaccinations multiples avant l’âge de 9 mois, c’est-à-dire pendant une période de la vie où les petits enfants sont en poussée de croissance majeure.
À partir de 1978 et jusqu’en 1996, il exerce la médecine à Orange dans le Vaucluse en tant qu’homéopathe, acupuncteur, et même psychanalyste aux alentours de 1995…
En 1994, Alain Scohy a découvre les microzymas et le Pr. Antoine Béchamp. Le travail remarquable du Pr Béchamp lui a permis de mieux comprendre les errements de la médecine officielle et de découvrir un nouveau paradigme médical plus cohérent en tenant compte des microzymas, des vitamines et de la psychosomatique.
En 1996, il est radié à vie de l’Ordre des Médecins pour délit d’opinion : il a osé demander à ses “pairs” l’application du principe de précaution et du Code de Déontologie Médicale lors de la campagne de vaccination contre l’hépatite B de 1994 dans les collèges et lycées : un vaccin nouveau, révolutionnaire, préparé sur des cellules OGM et testé à grands frais sur nos enfants. Dans cette affaire, le Conseil de l’Ordre des médecins était à la fois le plaignant et le juge !
À la suite de cette sanction, et avant qu’elle ne soit effective, il préfère démissionner et continue à travailler comme “accompagnateur-thérapeutes”. Écrivain et conférencier dans le Gard jusqu’en novembre 2002, il est obligé de s’expatrier suite à un redressement fiscal et un acharnement judiciaire. Il subira même quelques années plus tard des attaques de la MIVILUDES qui est la Mission interministérielle de vigilance et de lutte contre les dérives sectaires.
La sanction de l’Ordre a eu au moins un avantage essentiel selon lui. Elle lui a permis de comprendre qu’il ne faisait pas partie de ces “gens-là” ! Mais surtout, elle a libéré son esprit de toute contrainte dogmatique. Il a donc pu faire de son métier de médecin un art et une science en allant puiser à la source auprès de véritables chercheurs et scientifiques :
• Le Dr SEMMELWEIS (hygiène, contagion mais surtout respect du malade),
• Le Pr. Antoine BECHAMP (les microzymas),
• Le Pr. Louis-Claude VINCENT (la Bioélectronique),
• Le Dr MASCHI (la nocivité des champs électromagnétiques),
• Le Dr HAMER (La Médecine Nouvelle – apportant la clef des cancers, leucémies, sida, infarctus etc…)
• Le Dr KALOKERINOS (la vitamine C et le problème de la mort subite du nouveau-né),
Ces nouvelles orientations lui ont permis de modifier ma trajectoire professionnelle. Il le dit humblement : “Je n’ai pas fait de grande “découverte” à mon niveau, mais j’ai pu faire la synthèse de toutes ces connaissances.” Aujourd’hui, il continue de pratiquer et de partager sa démarche thérapeutique avec tous ses nouveaux acquis.
Combien de générations se sont vu interdire d’avoir des relations sexuelles normales parce que quelques illuminés ont réussi à transformer un syndrome qu’ils ont inventé de toute pièce en une MST mortelle ? Combien de malheureux souffrant de maladies diverses bien connues ont été envoyés ad-patres par des traitements (très onéreux) nouvellement créés dans le but déclaré de lutter contre ce syndrome virtuel ?
Avec cette du docteur Avec cette interview du docteur Claus Köhnlein, vous saurez tout sur cette immense arnaque du Sida qui dure depuis plus de 40 ans et qui préfigurait ce qui se passe aujourd’hui.
Claus Köhnlein : médecin spécialiste des maladies internes. A fait son internat dans le service d’oncologie de l’université de Kiel. Depuis 1993, travaille dans son propre cabinet médical, traitant les patients atteints d’hépatite C et du SIDA qui sont sceptiques à l’égard des médicaments antiviraux.
Vous êtes le co-auteur de Virus Mania, désormais également disponible en français, un livre que je recommande vivement. Comme la grande majorité jusqu’en mars 2020, je croyais aux virus, car cela équivalait à une croyance, puisque je n’avais jamais vérifié les expériences sur lesquelles la thèse était basée. Seulement, étant quelqu’un de très proche du monde réel, et non du monde fictif d’internet, je ne pouvais pas prendre au sérieux la possibilité d’un virus tueur prêt à bondir sur tout le monde ? Comment le pourrait-il puisque ce n’est pas censé être un organisme vivant. Et mes sens me disaient qu’il n’y avait rien de fâcheux, aucune masse de morts ou de maladies autour de moi. Votre livre, que j’ai lu en mars 2020, a commencé à m’ouvrir les yeux sur le plus grand mensonge au nom de la science, l’attribution aux virus des maladies. Il est extrêmement instructif, et j’ai été sidéré. Comme vous exhortez le lecteur, à vérifier vos références, à ne pas vous croire sur parole. C’est ce que j’ai fait, et c’était comme entrer dans une boîte de Pandore.
Lorsque les intérêts naturels et humains empiètent les uns sur les autres et que la sur-réglementation perturbe notre équilibre biologique, d’importantes questions se posent. Appartenons-nous à la nature ou la nature nous appartient-elle ?
Une enquête qui pousse à la réflexion, dans laquelle le documentariste Marijn Poels explore le besoin humain de contrôler notre climat, notre sécurité et de préférence, l’autre. La ligne de démarcation entre réglementation et manipulation est très mince. Lorsque la technologie règne en maître et que le bon sens s’évapore à l’épreuve du temps, l’humanité est sur le point de devenir l’outil.
À des kilomètres de la panique collective, de la peur et du chaos, il y a de l’espoir, de l’inspiration et une reconnexion.
Un excellent documentaire sur l’importance de la santé des sols dans l’environnement, du rôle des institutions tel que les Nations unies, des limites de la science, et des initiatives locales visant à redonner vie aux écosystèmes.
Un parallèle évident à faire avec notre propre terrain biologique et notre approche de la santé.
![Terrain Le Film – Documentaire Complet Partie 1 et 2 [VOSTFR]](/wp-content/uploads/2022/02/Terrain-le-film-cover2-1024x536.jpg)